
Artificial Intelligence and Machine Learning
AI Revolution of Materials Discovery
Principal Investigator:
Prof. Dr. Miguel Marques
Affiliation:
Ruhr Universität Bochum, Germany
Local Project ID:
pn25co
HPC Platform used:
SuperMUC-NG PH1-CPU at LRZ
Date published:
Introduction
Over the last few decades, ab-initio methods such as density-functional theory have become sufficiently accurate to allow for the prediction of many properties of new crystal structures. However, these predictions come at a significant cost, and due to the vastness of the space of possible materials, theoretical material discovery remains one of the most challenging questions in materials science. Machine learning methods, trained on existing databases of ab-initio calculations, have the potential to massively accelerate the process of theoretical materials discovery. One of the most important properties targeted is thermodynamic stability, which is used as a proxy to estimate the probability that a given compound can be synthesized.
Recently, we developed a neural network architecture specialized for predicting new stable materials, known as crystal graph attention networks. We used this network to predict the stability of around 15 billion compounds. The most promising compounds were then investigated with ab-initio theory in SuperMUC-NG, and added to our materials database alexandria [1].
Alexandria has grown over the past years to become the largest freely available database of computed properties of inorganic solids. It now includes entries for more than 4.4 million compounds, making it the largest (academic) database in the world. For comparison, AFLOW has around 3.5 million entries, the open quantum materials database has 1 million, and the materials project 155 thousand. Furthermore, alexandria combines this with a database of more than 140 thousand two-dimensional materials, 13 thousand one-dimensional materials, and 430 thousand entries calculated with a higher-accuracy method. Finally, all this information is available with a Creative Commons Attribution 4.0 License, that allows the sharing and adaptation of the data, and does not restrict any kind of academic or commercial use.
The effectiveness of alexandria relies on the abundance, diversity, and richness of crystal information, emphasizing the importance of size, structural diversity, and inclusion of computed properties for training robust machine-learning models in the field of materials science.
Results and Methods
In our workflow, we use our crystal-graph network model to predict the stability of a compound. This is measured by the energy distance to the so-called convex hull of thermodynamic stability, a hypersurface in the 100-dimensional materials space whose vertices are the stable compounds in the thermodynamic sense. A large distance indicates that the compound is highly unstable and is therefore of no practical use. A small distance, or if the compound is on the hull (zero distance) indicates stability towards decomposition, and a higher probability that the compounds can be synthesized experimentally. Factoring in errors in the theoretical framework and other physical effects such as temperature, defects, etc. one commonly uses a threshold of around 0.05-0.10 eV to distinguish the region of interesting compounds.
In the first step of the workflow, we select a known crystal structure that defines a “prototype”. This prototype specifies the crystal symmetry, the crystal unit cell, and the positions of the atoms in the cell. We then use a combinatorial engine to transmute the atoms in this structure into all other possible chemical elements. For example, the rocksalt crystal structure of the common NaCl kitchen salt will be transmuted in NaF, or LiCl (also naturally occurring salts) but also in RbU, TcH, and all other combinations. The neural network then decides if this is a viable compound, and, if so, passes it to the following stage.
The following stage consists of an ab initio calculation, using density functional theory as implemented in the code vasp, to calculate the basic properties of the proposed compound – and validate the stability prediction of the neural network. For each material we retain around 10 (text) files that contain the most output of the run. These are then compressed and transferred to our university where they are stored.
A calculation with vasp is around 1 million times slower than with the neural network, and it is in this step that the computational resources provided by SuperMUC-NG are essential. The compound is then screened for interesting properties (electronic, magnetic, mechanical, etc.) and inserted into the alexandria database. In the last two years we have used nearly 100 million core-hours in SuperMUC-NG, which allowed us to compute close to two million compounds. The process scales linearly with the number of compounds allowing us to make efficient use of the machine.
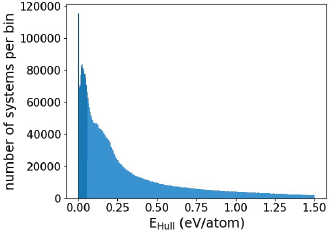
Figure 1: Distribution of the distance to the convex hull of thermodynamic stability of the compounds in alexandria. Only the compounds that are close to the hull (indicated by a darker shade of blue) have a high probability of being synthesized experimentally.
Of course, more data usually means better machine learning models, therefore after each “round” of vasp calculations we retrain the neural network to improve its accuracy. The improvement in the prediction of stable, or near stable compounds, can be seen in Figure 2. There we show the distribution of the distance to the convex hull of thermodynamic stability for three consecutive rounds. It is clear that in round 3 a much larger percentage of compounds are close to thermodynamic stability due to the improved training of the crystal graph network allowed by the extra data.
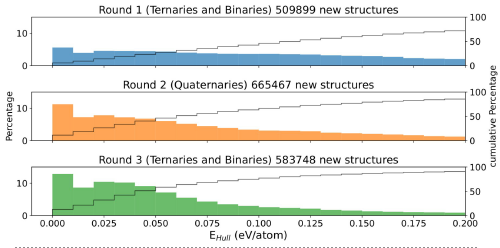
Figure 2: Distribution of the distance to the convex hull for the compounds calculated with vasp in three consecutive “rounds”. Between each round, the neural network that proposes the materials to be calculated is retrained with all data from the previous rounds.
Besides allowing for better training of machine learning models, any of these newly discovered compounds may hold the key to important technological advances. For example, recently we scanned the compounds in alexandria for high-temperature conventional superconductors. The search yielded some surprising results, such as the compound LiMoN2 that we predict to superconduct below 40 K, or the hydride Mg2IrH6 that, if synthesized, should be a superconductor at ambient pressure at temperatures surpassing the boiling point of nitrogen.
Ongoing Research/Outlook
At the moment, and in spite of the incredible advances of the past years, we only know a very small part of material space. As such, we are continuing this project in SuperMUC-NG to unveil more stable compounds with potential technological applications.
References and Links
[2] J. Schmidt, N. Hoffmann, H.-C. Wang, P. Borlido, P.J.M.A
[3] J. Schmidt, H. Wang, G. Schmidt, and M. Marques, npj Comput. Mater. 9, 63 (2023).
[4] T.F.T. Cerqueira, A. Sanna, M.A.L. Marques, Adv. Mater. 36, 2307085 (2024).
[5] A. Sanna, T.F.T. Cerqueira, Y.-W. Fang, I. Errea, A. Ludwig, M.A.L. Marques, npj Comput Mater. 10, 44 (2024).