MATERIALS SCIENCE AND CHEMISTRY

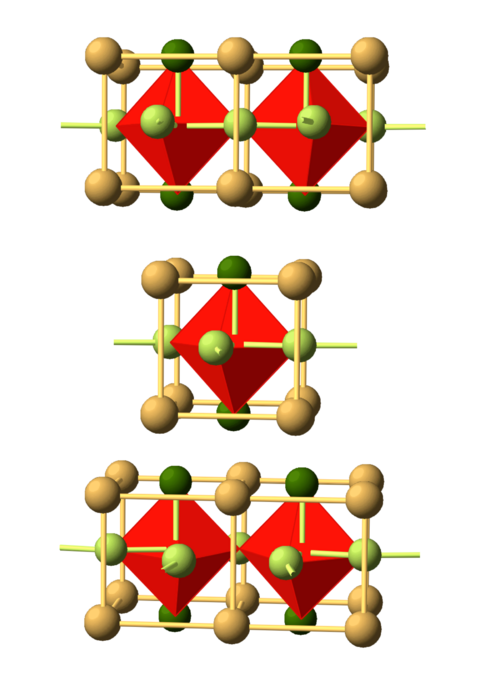
Materials Science and Chemistry

Materials Science and Chemistry
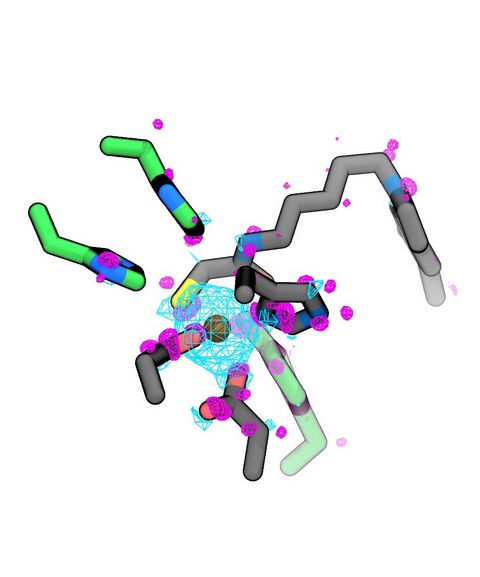
Materials Science and Chemistry
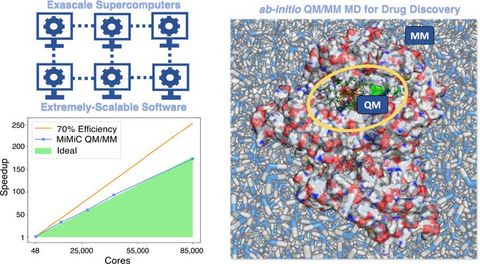
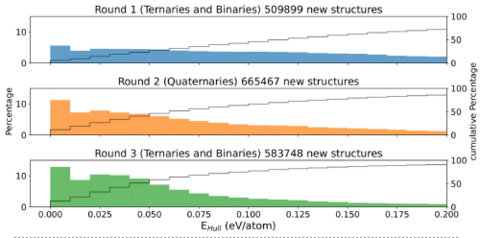
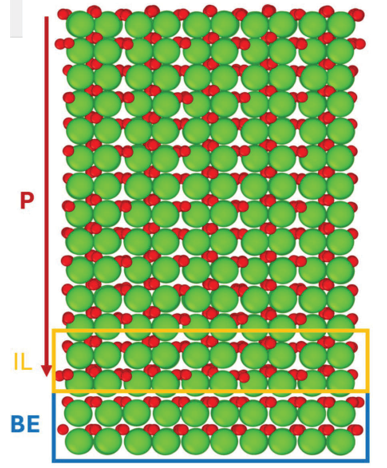
Materials Science and Chemistry
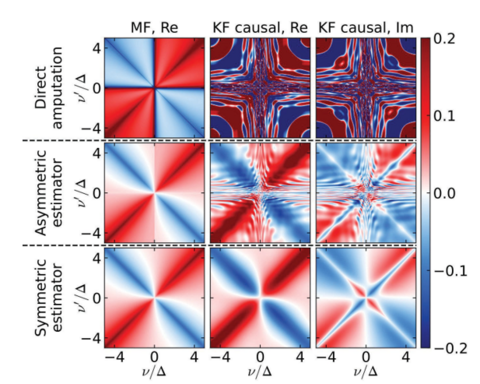
Materials Science and Chemistry
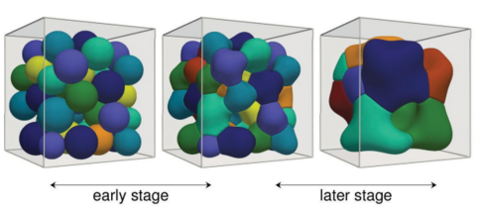
Materials Science and Chemistry
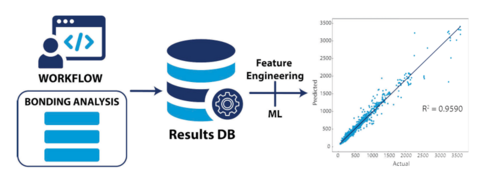

Materials Science and Chemistry
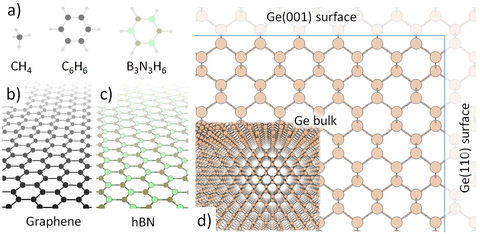

Materials Science and Chemistry
Materials Science and Chemistry
Materials Science and Chemistry
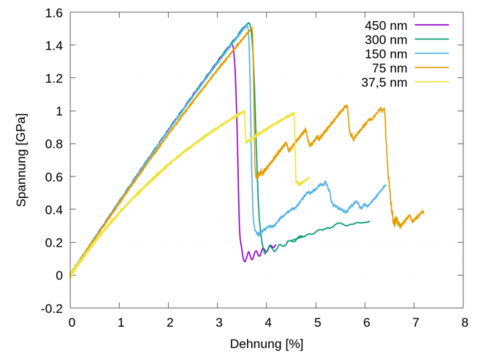
Materials Science and Chemistry
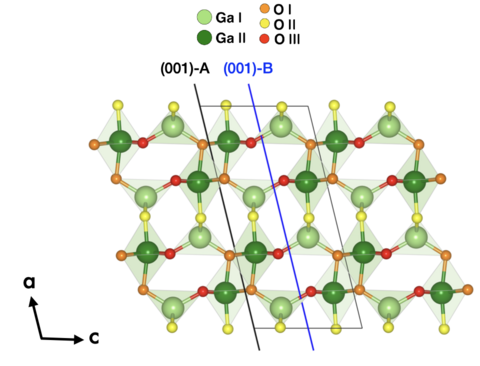
Materials Science and Chemistry
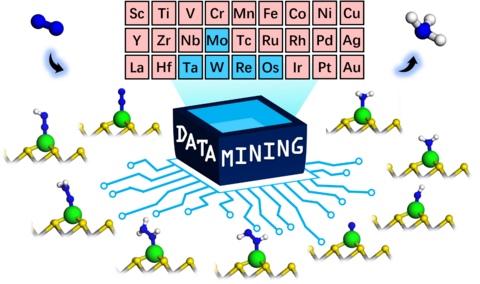
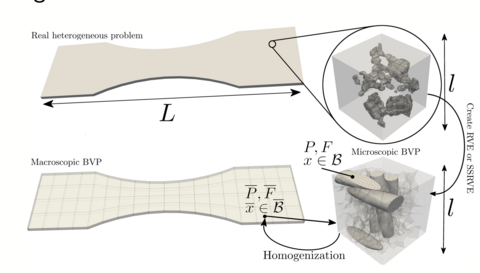
Materials Science and Chemistry
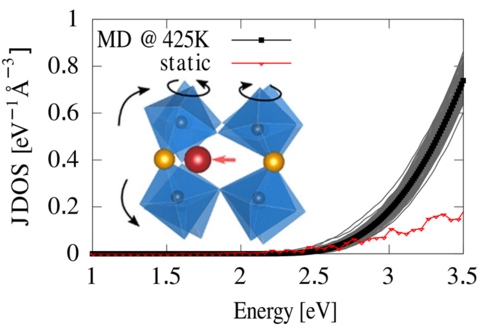
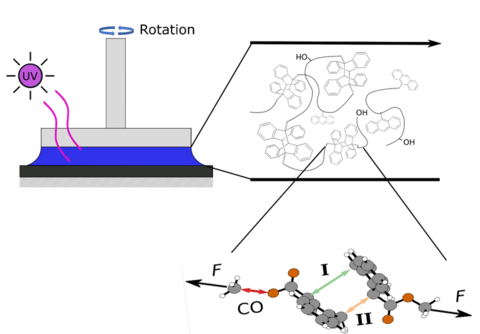
Materials Science and Chemistry

Materials Science and Chemistry

Materials Science and Chemistry

Materials Science and Chemistry
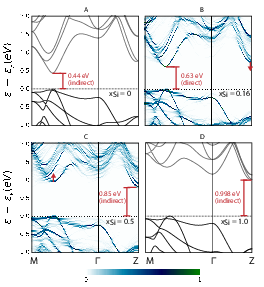
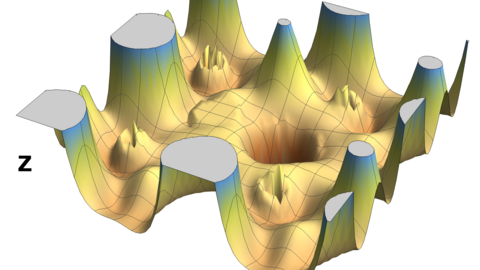
Materials Science and Chemistry
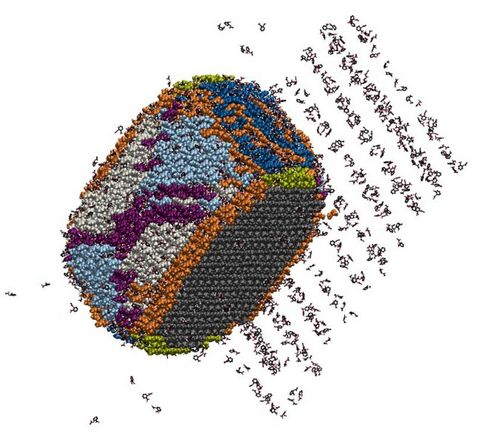
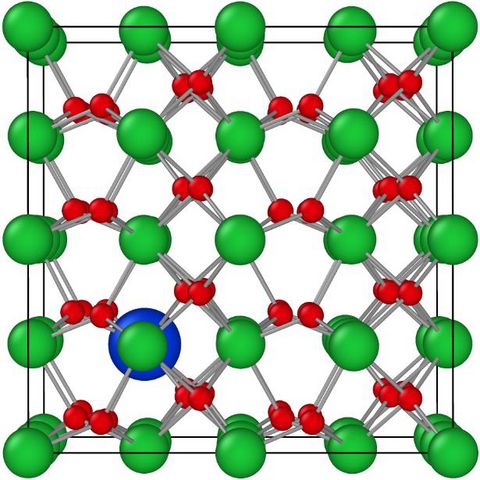

Materials Science and Chemistry
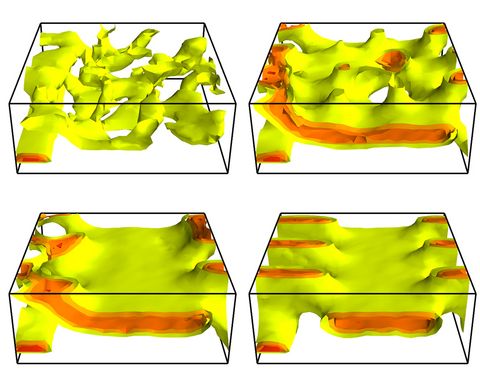
Materials Science and Chemistry
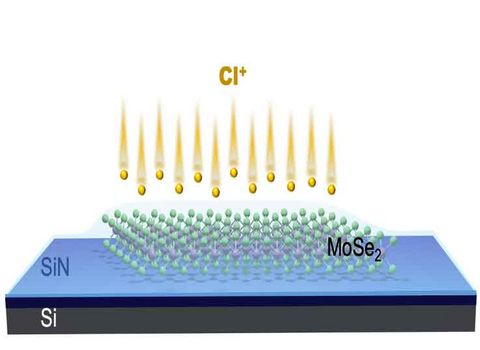
Materials Science and Chemistry
Materials Science and Chemistry
Materials Science and Chemistry

Materials Science and Chemistry
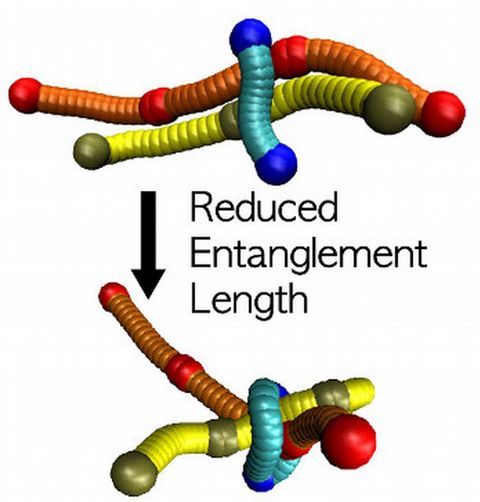
Materials Science and Chemistry
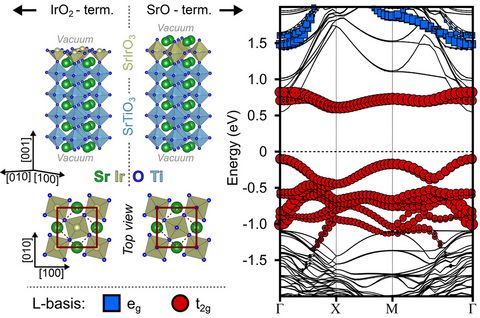
Materials Science and Chemistry
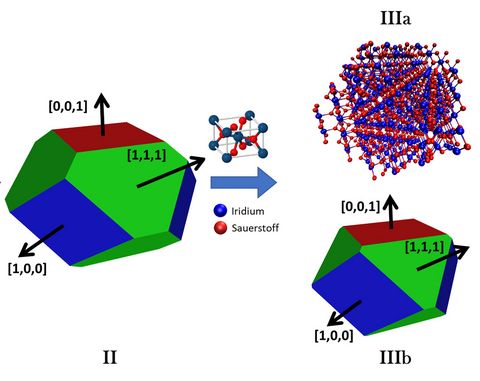
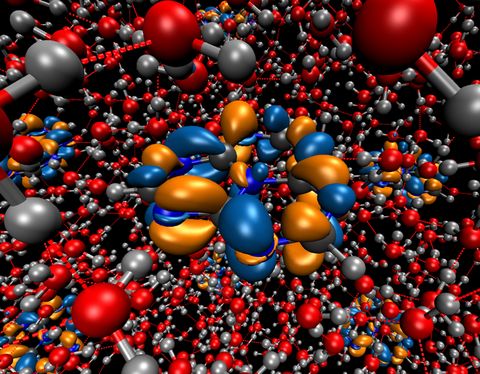
Materials Science and Chemistry
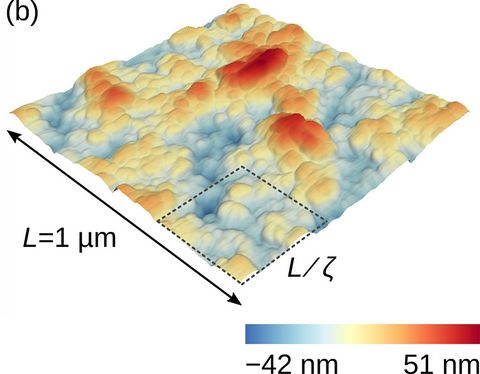
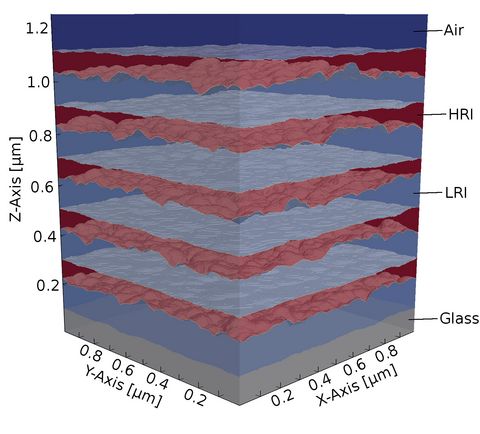
Materials Science and Chemistry
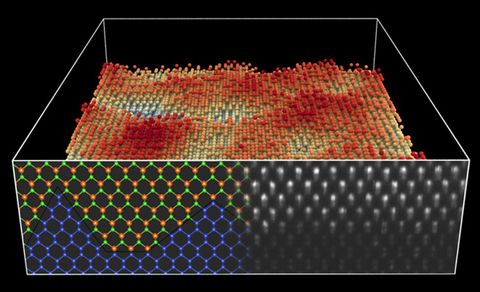
Materials Science and Chemistry
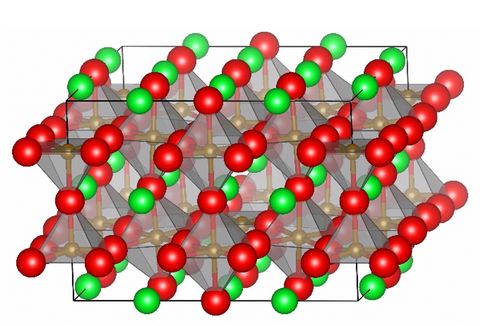
Materials Science and Chemistry
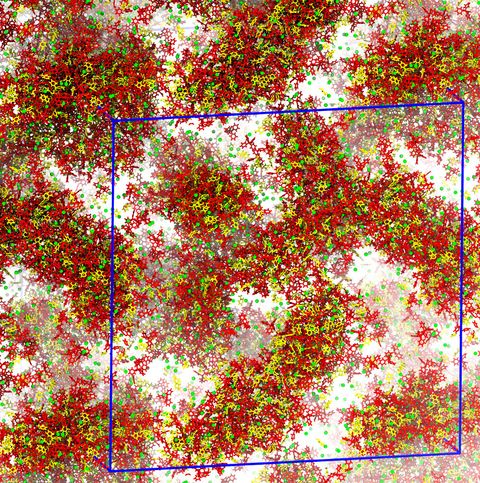
Materials Science and Chemistry
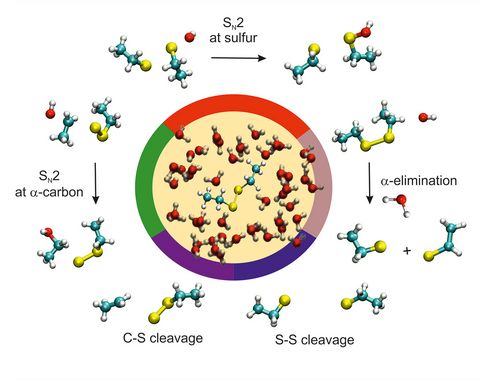
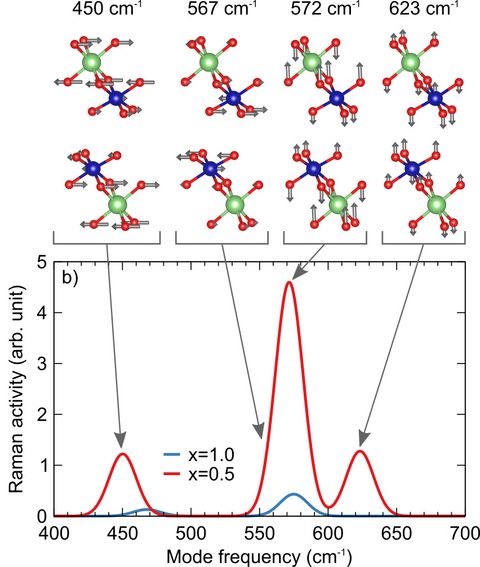
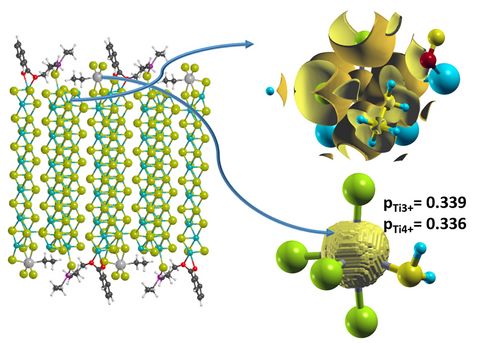
Materials Science and Chemistry
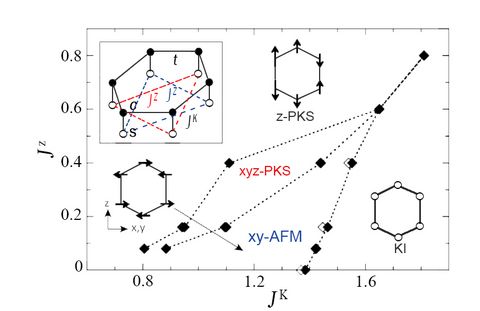
Materials Science and Chemistry

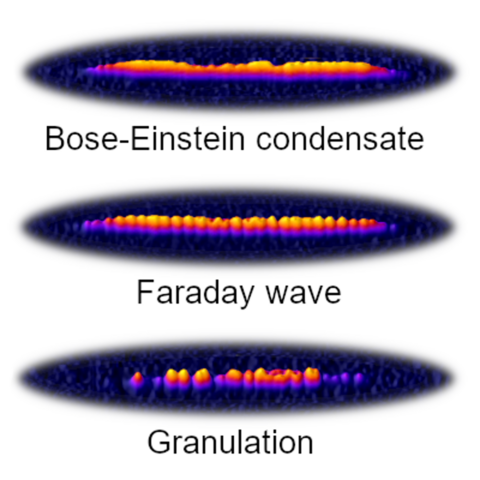
Materials Science and Chemistry
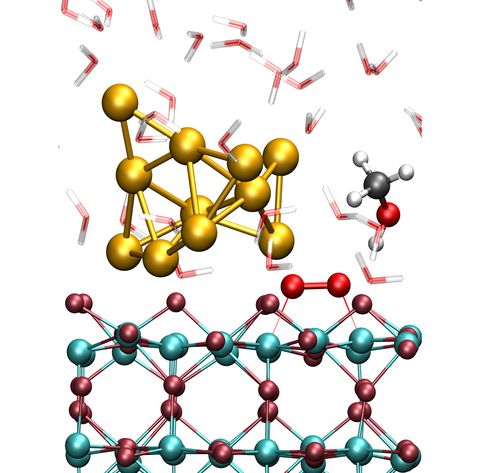
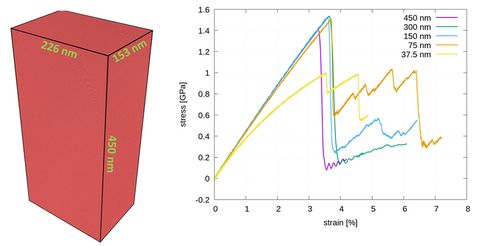
Materials Science and Chemistry
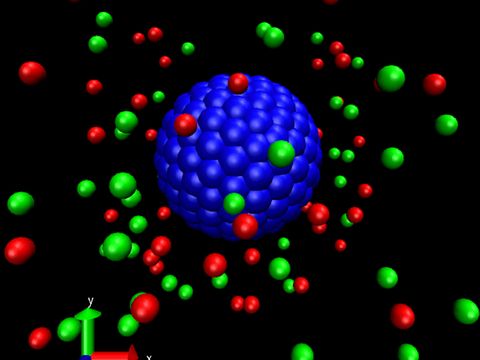
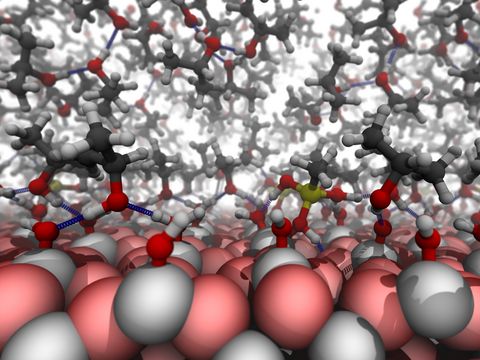
Materials Science and Chemistry
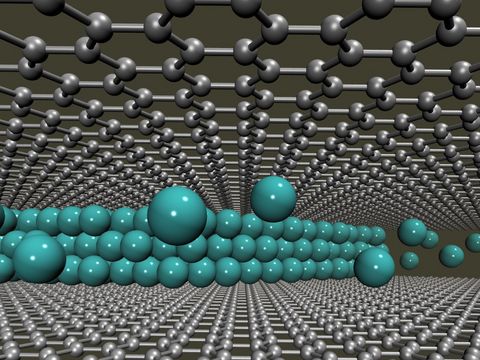
Materials Science and Chemistry
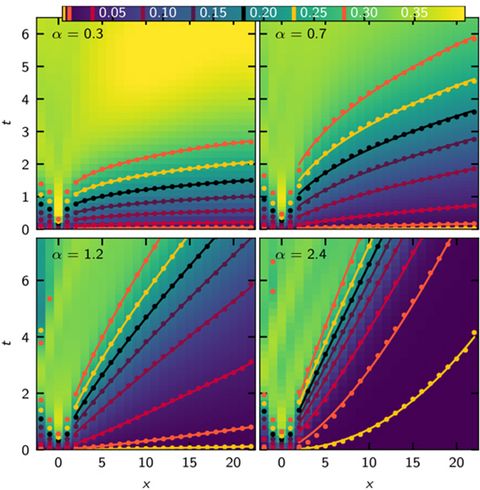
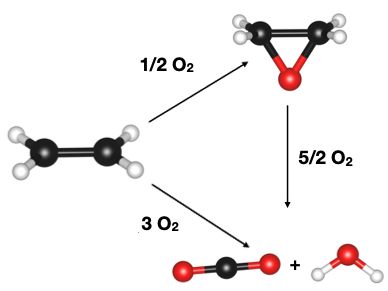
Materials Science and Chemistry
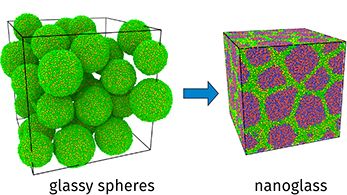
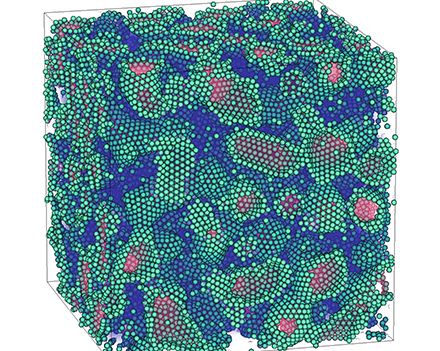
Materials Science and Chemistry
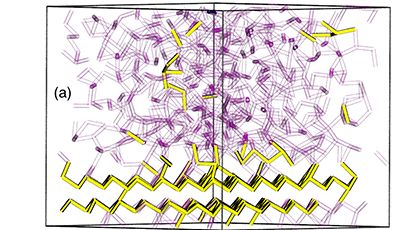
Materials Science and Chemistry
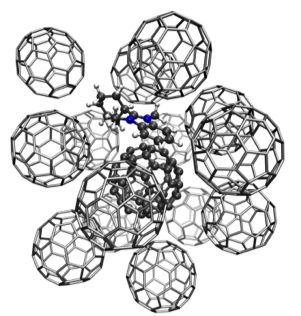
Materials Science and Chemistry
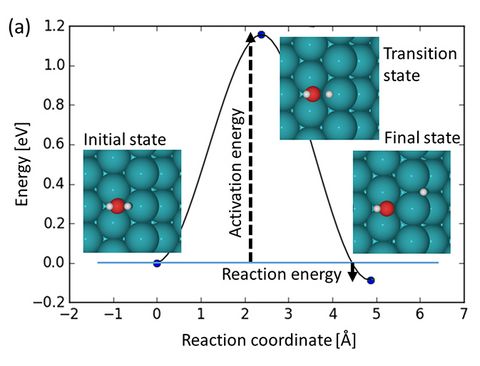
Materials Science and Chemistry
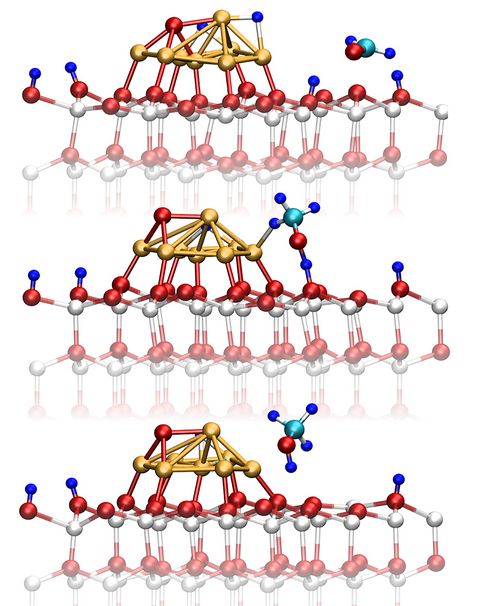
Materials Science and Chemistry

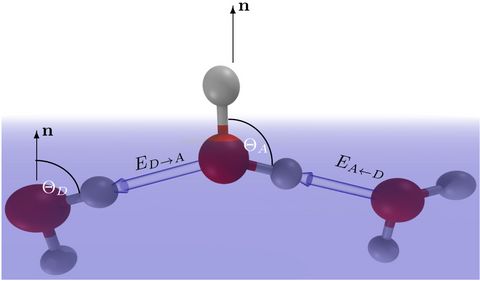
Materials Science and Chemistry
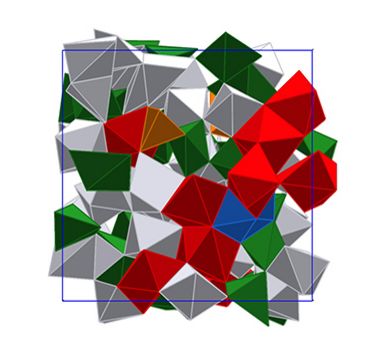
Materials Science and Chemistry
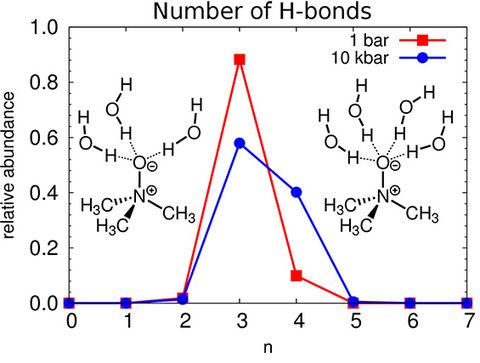
Materials Science and Chemistry

Materials Science and Chemistry
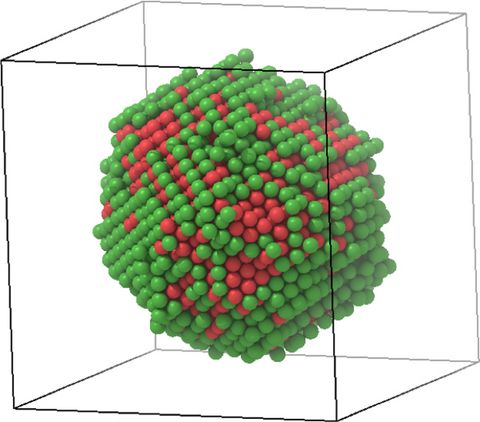
Materials Science and Chemistry
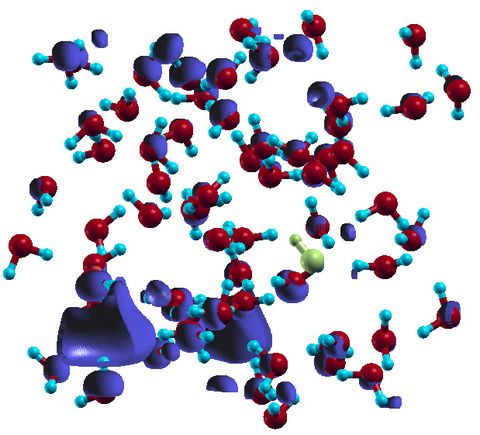
Materials Science and Chemistry
Materials Science and Chemistry
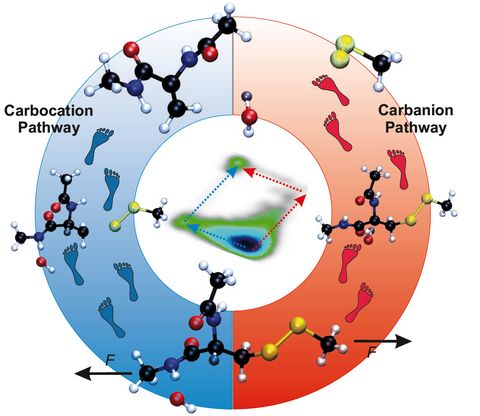
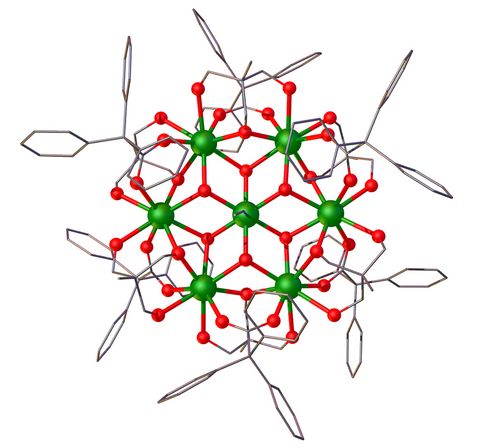
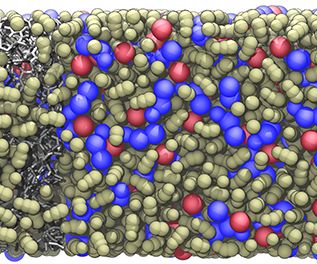
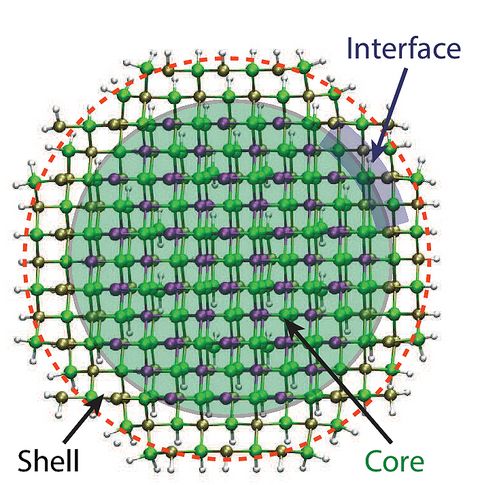
Materials Science and Chemistry
Materials Science and Chemistry

Materials Science and Chemistry
Materials Science and Chemistry

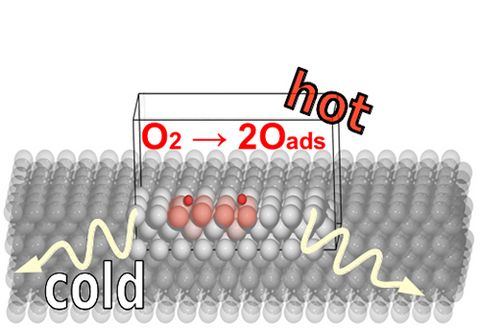
Materials Science and Chemistry
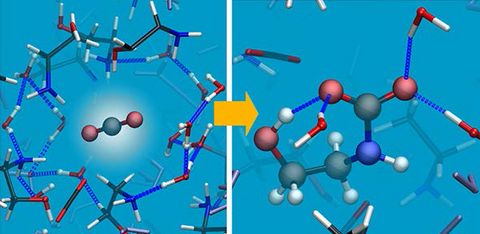
Materials Science and Chemistry

Materials Science and Chemistry
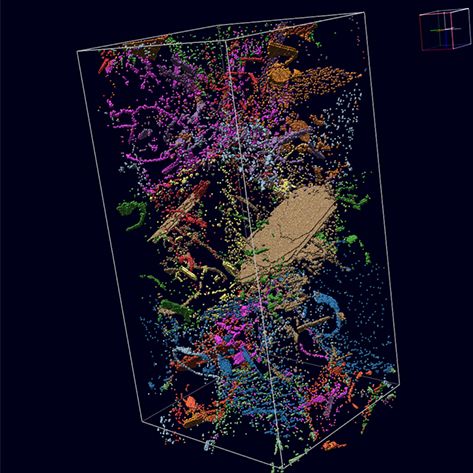
Materials Science and Chemistry
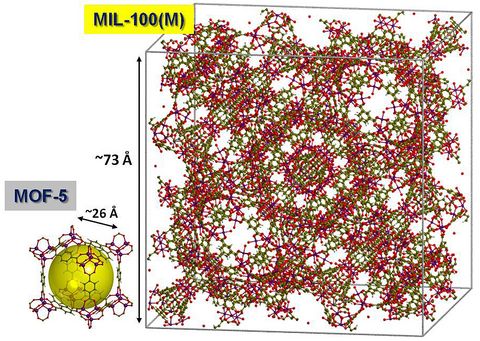
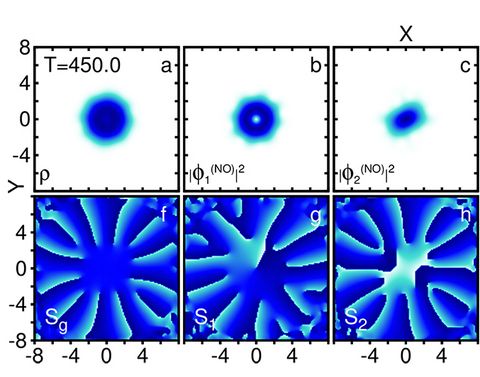

Materials Science and Chemistry
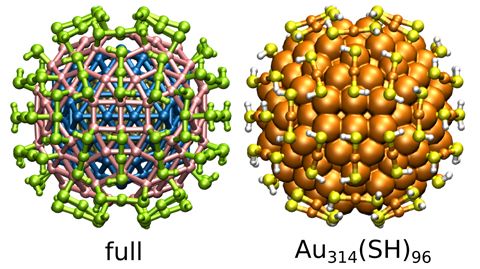
Materials Science and Chemistry
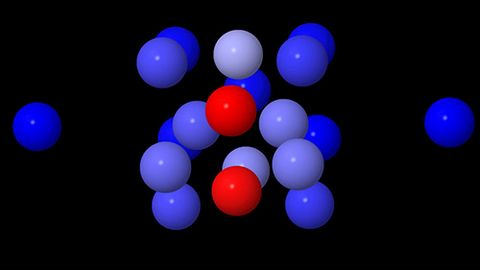
Materials Science and Chemistry

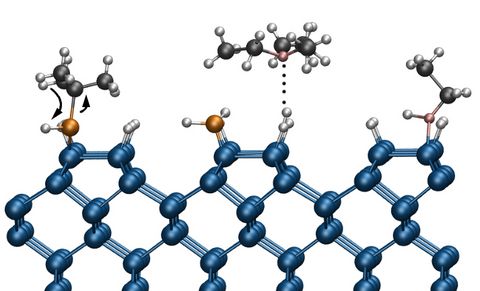
Materials Science and Chemistry

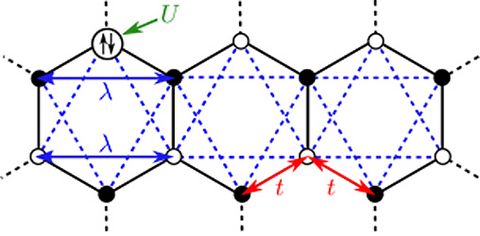
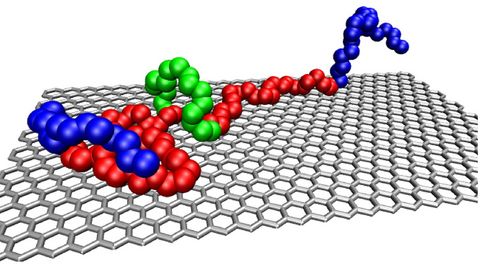

Materials Science and Chemistry