
MATERIALS SCIENCE AND CHEMISTRY
Linear-Scaling Transport Approach for Innovative Electronic Materials
Principal Investigator:
Frank Ortmann
Affiliation:
Technische Universität Dresden (Germany)
Local Project ID:
pr84po
HPC Platform used:
SuperMUC (LRZ)
Date published:
Researchers elucidate the molecular doping of prototypical representatives for the class of molecular semiconductors. As n-type dopants, molecular radicals, closed-shell molecules and metal-organic species are compared. By using the HPC system SuperMUC, they simulate doping-induced states and compare the simulations with ultraviolet photoemission and inverse photoemission spectra. One challenge in the simulations is the necessary accuracy of the computation of the involved energies in the doping process, which requires ab initio approaches. In addition, the disordered material blends include many complex molecules whose charging states and charging energies need to be simulated by taking into account the blend’s dielectric properties and microscopic molecular arrangement.
Introduction
Semiconductors are a class of materials with unique electrical properties. Their charge transport can be specifically influenced by doping, whereby electron mobilities in conventional semiconductors can be changed by many orders of magnitude. This functionality is indispensable in the modern semiconductor industry. The same applies to organic semiconductors, for which doping has been studied in the context of various applications. Here, molecular dopants are introduced into molecular host materials that behave qualitatively and quantitatively different from inorganic semiconductors. The more complicated organic systems pose great challenges with unanswered questions regarding their microscopic description [1].
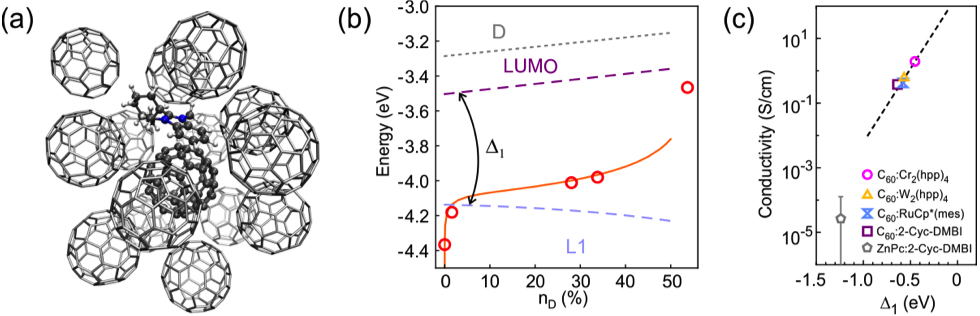
Figure 1: Structure, Electronic Properties and Charge Transport. (a) Geometry of molecular cluster consisting of a benzimidazoline (DMBI) dopant and a C60 molecule (ball and stick model; atoms are indicated in blue (N), dark grey (C) and light grey (H)) surrounded by C60molecules (stick model). (b) Comparison of the Fermi level shift between theory (orange line) and experiment (circles) at room temperature. The C60 states LUMO (energy EA(C600)) and L1 (energy IPD+(C60-)) are indicated as dashed lines. The dotted line (indicated by ‘D’) is the LUMO of the dopant cation. (c) Conductivities in n-doped C60 at doping concentration nD=2% versus the characteristic binding energy Δ1. The dashed line is a guide to the eye for the C60host material. Figure reproduced after C. Gaul et al. Nature Materials 17, 439–444 (2018).
Results
In this project, the researchers elucidate the molecular doping of organic semiconductors for C60-fullerene and Zinc-phthalocyanine. Both are prototypical representatives for the class of molecular semiconductors. As n-type dopants, we compare molecular radicals, closed shell molecules and metal-organic species. We study, by using the HPC system SuperMUC at the Leibniz Supercomputing Centre in Garching (LRZ), doping-induced states and simulate ultraviolet photoemission and inverse photoemission spectra. One of the challenges in the simulations is the necessary accuracy of the computation of the involved energies in the doping process, which requires ab initio approaches. In addition, the disordered material blends include many complex molecules whose charging states and charging energies need to be simulated by taking into account the blend’s dielectric properties and microscopic molecular arrangement.
The group of Dr. Frank Ortmann from the Center for Advancing Electronics Dresden of the Technische Universität Dresden has advanced the microscopic understanding of doping in organic materials [2]. The comprehensive simulations allow for an accurate description of doping-induced gap states and their energies for the n-doped systems under study. We unveil their role in determining the position of the Fermi energy. In addition, we demonstrate a systematic relationship between the energy of these gap states (characteristic binding energy Δ1) and the electrical conductivity, which allows estimating the conductivity from a simple energetic parameter. Our theoretical predictions have been validated by comprehensive experimental studies together with our collaboration partners.
References:
[1] K. Walzer, B. Maennig, M. Pfeiffer and K. Leo “Highly efficient organic devices based on electrically doped transport layers” Chem. Rev. 107, 1233–1271 (2007).
[2] C. Gaul, S. Hutsch, M. Schwarze, K. S. Schellhammer, F. Bussolotti, S. Kera, G. Cuniberti, K. Leo and F. Ortmann “Insight into doping efficiency of organic semiconductors from the analysis of the density of states in n-doped C60 and ZnPc” Nature Materials 17, 439–444 (2018).
Scientific Contact:
Dr. Frank Ortmann
Technische Universität Dresden
Center for Advancing Electronics Dresden
D-01062 Dresden (Germany)
E-mail: frank.ortmann [at] tu-dresden.de
June 2018