Principal Investigator:
Dominik Marx
, Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum (Germany)
HPC Platform used:
JUQUEEN of JSC
Local Project ID:
hbo27
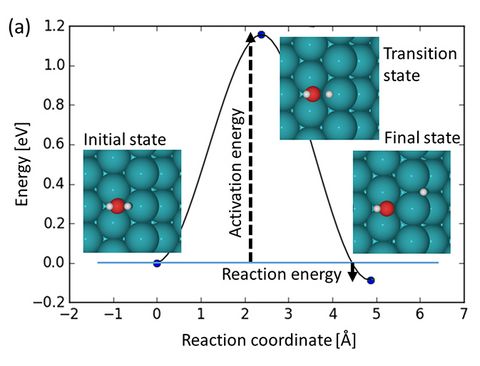
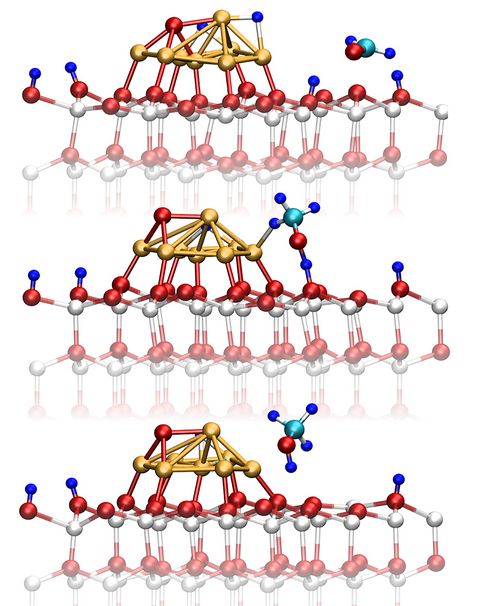
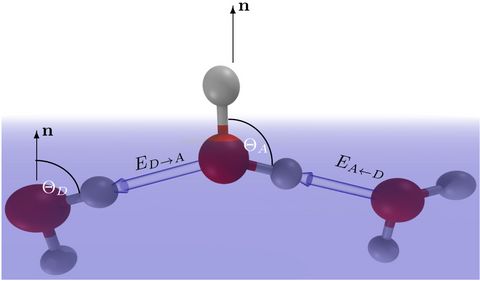
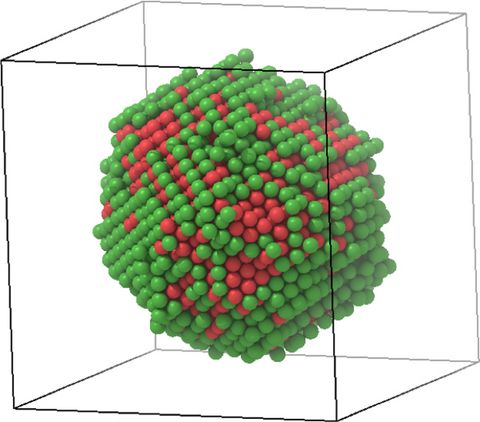
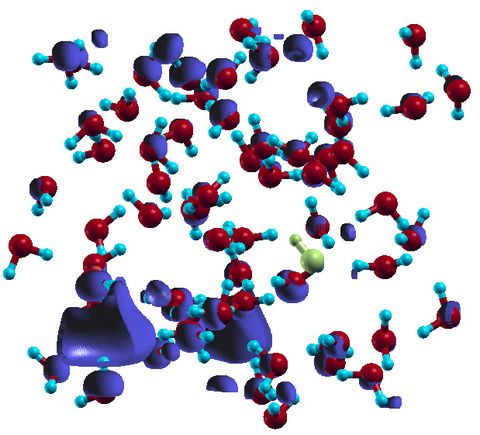
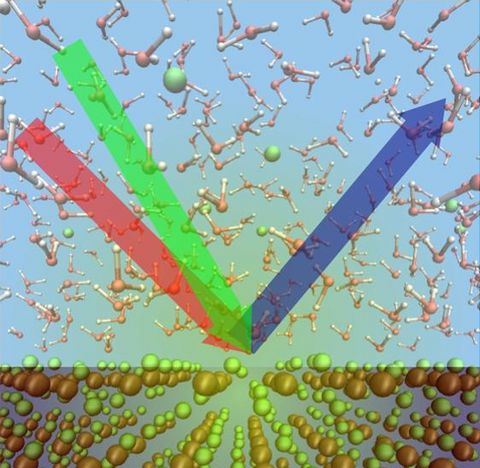
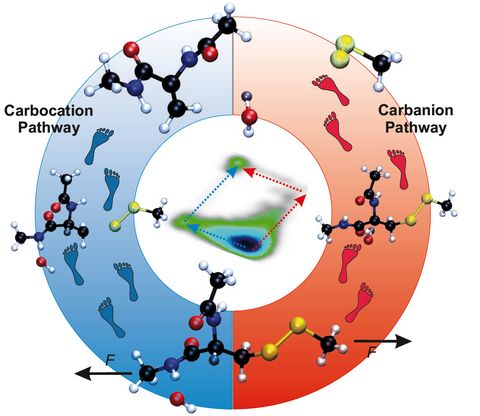
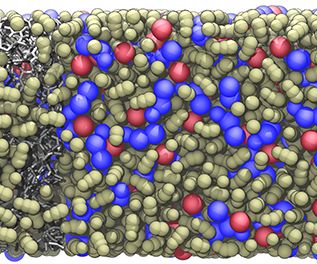
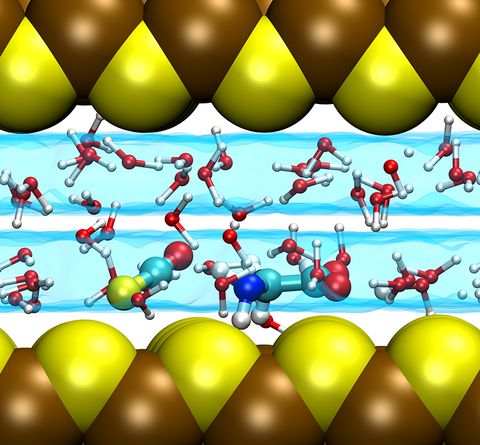
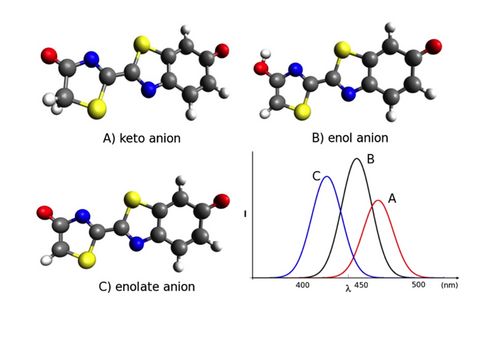
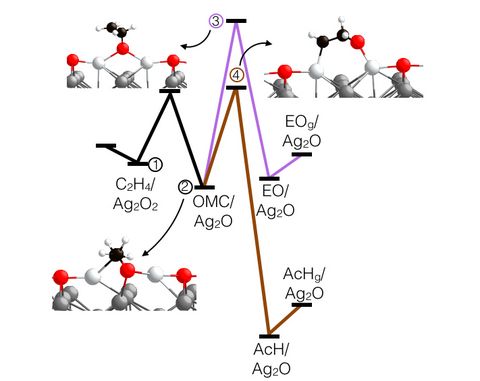
Prebiotic Chemistry is the study of those chemical reactions that could have taken place on the early Earth by which, starting from small molecules like H2O, NH3, CO2, SH2 or simple amino acids, more complex molecules were formed. This leads eventually to the formation of biomacromolecules as we know them from today's life, for instance proteins, RNA or DNA but also lipids. Advanced computer simulations in conjunction with large-scale HPC facilities and scalable codes allow one to investigate at the very molecular level not only how these reactions could have happened, but more importantly how they are affected by factors like temperature, pressure, or the presence of mineral surfaces to name but a few.