
MATERIALS SCIENCE AND CHEMISTRY
On the Liquid Mechanism of Methanol Oxidation at Au/TiO2 Nanocatalysts
Principal Investigator:
Dominik Marx
Affiliation:
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum (Germany)
Local Project ID:
pr74va
HPC Platform used:
SuperMUC of LRZ
Date published:
Introduction
The backbone of industrial chemistry is heterogeneous catalysis. Even small improvements of the used catalyst, often based on mechanistical insights, may enhance the efficiency in terms of energy consumption and/or selectivity. Such insights can be provided by the ‘Solvation Science approach’ to theoretical heterogeneous catalysis in the liquid phase, while experiments often lack the required atomistic resolution.
Highly dispersed gold/titania catalysts are widely used for key reactions, notably including the selective oxidation of alcohols in the liquid phase. The mechanistic details of this reaction are mostly unknown, while the corresponding gas-phase process is often investigated. The pivotal role of water in stabilizing charge transfer and its actual chemical role in the reaction mechanism remains poorly known.
Here, we take advantage of enhanced sampling ab initio molecular dynamics (AIMD) [1] simulations using a well-established Au/TiO2 nanocatalyst model [2, 3, 4] (Figure 1) in order to elucidate the mechanistic details of thermally activated liquid-phase methanol oxidation at elevated T and p in accordance with experimental conditions.
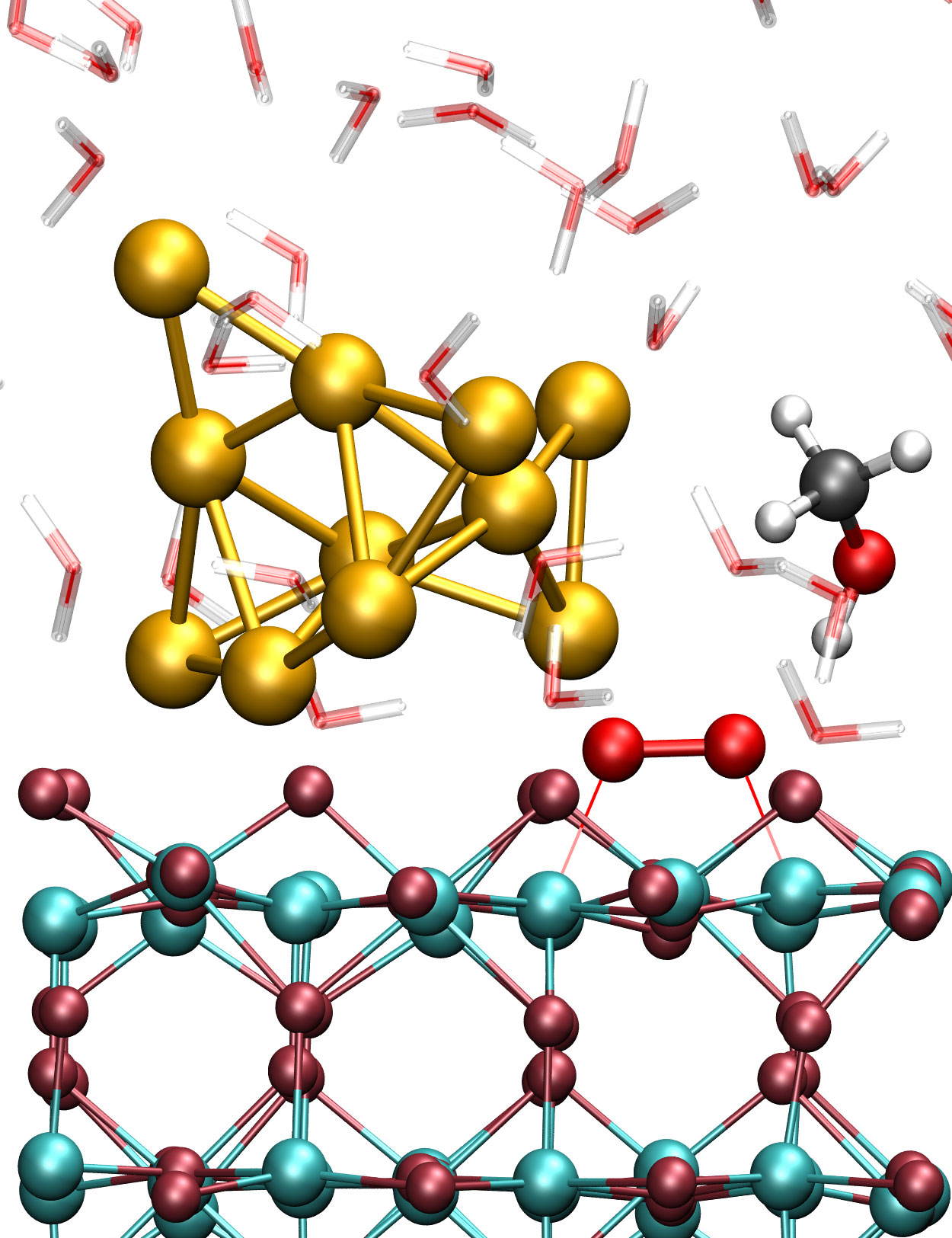
Figure 1: Close-up of the liquid phase Au/TiO2 nanocatalyst system used to simulate methanol oxidation by dioxygen that is activated at the AuNP/titania perimeter site. The TiO2 slab is shown in dark red (O) and cyan (Ti), the AuNP in gold, the adsorbed O2 in red, the CH3OH in black (C), red (O) and white (H), and the H2O molecules in transparent red (O) and white (H).
Copyright: RUB, GermanyResults and Methods
We performed large-scale multiple walker ab initio metadynamics simulations with the Au nanoparticle (AuNP) being in contact with the dioxygen and methanol reactants in the gas phase as well as in neutral water [5]. In the gas phase, the methanol molecule is adsorbed via weak interactions of its OH group with the O2 (see structure aG in Figure 2). In the course of the reaction, one of the aliphatic hydrogens moves close enough to the AuNP, leading to C-H activation (see bG in Figure 2). The final oxidation of methanol is observed exclusively if the O2 molecule dissociates. The dissociation is caused via charge transfer from the AuNP resulting in a partially positive AuNP and a partially negative O2 species. After dissociation, the protonic hydroxyl’ H and the hydridic aliphatic H are respectively transferred to the O2 and the AuNP in a concerted fashion.
In liquid water charged species can be stabilized thanks to its high dielectric constant, which allows a stepwise mechanism for methanol oxidation. The reaction starts with the O2 dissociation due to charge transfer from the AuNP, which carries more charge than in the gas phase due to the charge donated from its solvation shell. After the dissociation, one of the nascent O atoms is able to accept the hydroxyl proton, which was observed to be transferred via a Grotthuss-like diffusion process involving a water molecule. The resulting methoxy intermediate is a stable intermediate in water. Once the aliphatic H atom of the methoxy is close enough to the AuNP, it is transferred as a hydride yielding the product formaldehyde.
In summary, the presence of water crucially changes the mechanism of methanol oxidation at Au/TiO2. Our ‘Solvation Science approach’ is able to determine the mechanistic changes of the reaction due to the presence of water, not only establishing the qualitatively different mechanism when comparing the liquid phase with the gas phase reference, but also rationalizing the changes in terms of the charge transfers occurring between the involved species.
Performance and scaling
The AIMD simulations were carried out using CPMD [6] which is a DFT-based molecular dynamics code. The electronic structure of the nanocatalyst model was described by a plane wave basis set in combination with ultrasoft pseudopotentials. The ionic and electronic degrees of freedom of the system are time-propagated with an extended Lagrangian scheme, which is the most computationally demanding part of the calculation.
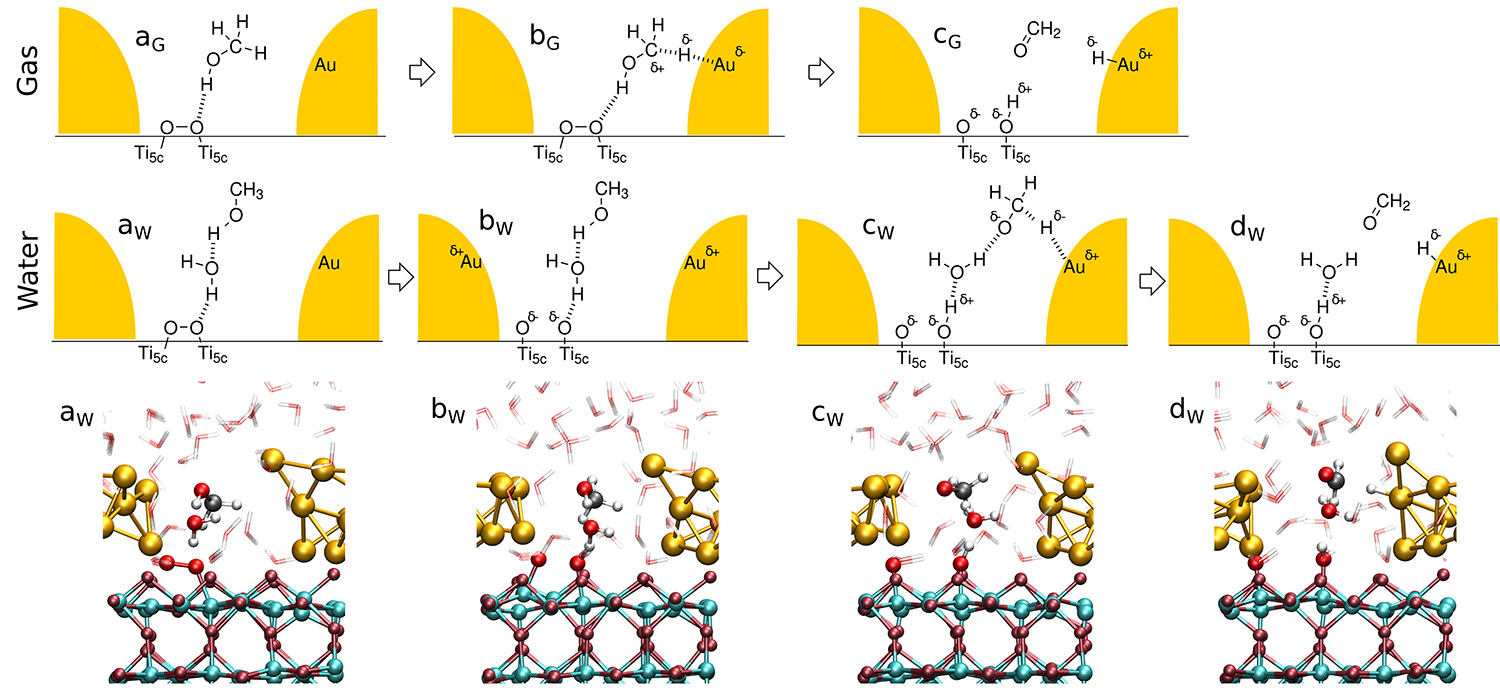
Figure 2: Top panels: Schematic reaction mechanisms for the CH3OH oxidation on O2/Au/TiO2 in the gas phase and in water as extracted from the ab initio simulations (the AuNP is depicted in gold; only the reactive water molecules involved in the liquid-phase mechanism are shown). Bottom panel: Real-space snapshots of the aqueous system, from left to right: structures aW, bW, cW and dW.
Copyright: RUB, GermanySuch task is efficiently performed using processor groups (Pgr). Within a Pgr the parallelization is realized via MPI and either MPI or OpenMP/Vector for inter-node and intra-node communication, respectively. At this level of parallelization the allocation of 160 cores allowed for the highest speedup for our Au/TiO2 nanocatalyst model. Herein the 3D-FFT of the electronic wavefunctions is split up most efficiently into one 2D-FFT per processor core. Further levels of parallelization within the CPMD code, e.g. via Kohn-Sham orbitals, resulted in lower speed-up. Thus, production runs were carried out with 2D-FFT parallelization giving the best use of the granted CPU time and the best time to solution.
On SuperMUC CPMD propagation of a single gas phase Au/TiO2 nanocatalyst model required 28.1 and 19.1 core·min per MD step on Phase 1 thin nodes and on Phase 2 nodes, respectively, fully employing 10 nodes on Phase 1 or 6 nodes on Phase 2. For the liquid phase 62.5 and 47.2 core·min per MD step on Phase 1 and on Phase 2 were required, respectively. There exists another level of parallelization which is the multiple walker extension to the metadynamics method. Each “walker” is a different replica of the system, and the different walkers interact in building up the biasing potential with a negligible communication effort. Hence, this algorithm shows a linear scaling with respect to the number of walkers, being possible to use tenths of walkers. On SuperMUC partitions the best performance between queue waiting vs. production was achieved using up to ten walkers, i.e. 1960 cores. On SuperMUC this project has used approximately ten million core hours. Besides the bookkeeping of the trajectory data at every MD step, a larger disk I/O demand during such jobs was the writing of complete restart files (each of 1.9 GiB in size) which was done every four hours. The maximum storage needed in the SCRATCH, WORK and PROJECT filesystems was 1 TiB, 3 TiB and 100 GiB, respectively.
On-going Research / Outlook
The simulations with our Au/TiO2 model on SuperMUC Phase 2 are speeded-up by about 40% compared to Phase 1. This allows for larger system sizes and ab initio molecular dynamics simulations with computationally more demanding electronic structure methods. Currently we are using an extended Au/TiO2 model to investigate the origin of the enhanced O2 activation at this catalyst, with which we will be able to quantify such activation enhancement in liquid vs. gas phase in terms of free energy barriers, also comparing different adsorption sites. Such insights will be of great interest for the heterogeneous catalysis field. These simulations require at least twice the computational resources of the present study. We estimate an increase in computational cost by about 100 times for upcoming projects which would be required and hopefully accessible by “SuperMUC Next Generation”.
References and Links
[1] D. Marx and J. Hutter, “Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods”, Cambridge University Press (2009)
[2] M. Farnesi Camellone, D. Marx. J. Phys. Chem. Lett. 4,514–518 (2013).
[3] M. Farnesi Camellone, P. M. Kowalski, D. Marx. Phys. Rev. B 84, 035413 (2011).
[4] M. Farnesi Camellone, D. Marx. J. Phys. Chem. C 118, 20989–21000 (2014).
[5] D. Muñoz-Santiburcio, M. Farnesi Camellone, D. Marx. Angew. Chem. Int. Ed. DOI:10.1002/anie.201710791
[6] CPMD, http://www.cpmd.org/
Research Team
Prof. Dr. Dominik Marx (PI), Dr. Daniel Muñoz-Santiburcio, Niklas Siemer (Ruhr-Universität Bochum, Germany)
Scientific Contact
Niklas Siemer
Lehrstuhl für Theoretische Chemie
Ruhr-Universität Bochum (RUB)
D-44780 Bochum (Germany)
e-mail: niklas.siemer [@] rub.de
https://www.theochem.rub.de
https://www.solvation.de
NOTE: This report was first published in the book "High Performance Computing in Science and Engineering – Garching/Munich 2018".
LRZ Project ID: pr74va
September 2019