
MATERIALS SCIENCE AND CHEMISTRY
Steering Complex Chemical Reactions by Mechanical Forces
Principal Investigator:
Dominik Marx
Affiliation:
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum (Germany)
Local Project ID:
hbo38
HPC Platform used:
JUQUEEN of JSC
Date published:
The Challenge
Bond breaking and making processes are key to chemisty. To overcome the energy barrier which separates the initial state (reactants) from the final state (products) of such chemical transformations one must pump energy into the molecular system – usually in the form of heat. But let us imagine that one could stretch the molecule using external forces in order to supply at least part of the required activation energy in terms of mechanical energy – instead of heating the system. Indeed, such mechanochemical experiments are nowadays possible in the laboratory using various techniques such as sonication of solutions or single-molecule force spectroscopy1. Computationally, covalent mechanochemistry requires the investigation of chemical reactions as a function of constant external forces that act on the molecule as it undergoes chemical transformations1.
The Question
Disulfide bonds are known for long to stabilize protein structures by imposing covalent cross-links. More recently they have been found to regulate protein activity as well by undergoing chemical reactions themselves. However, the chemistry of disulfide bond cleavage reactions is astonishingly rich and includes also β–elimination reactions in alkaline solution instead of the usual nucleophilic substitution at one of the sulfur atoms. In a GCS Large–Scale Project of the Gauss Centre for Supercomputing, we studied computationally both reaction channels as a function of increasingly large mechanical forces using a carefully selected disulfide model system in alkaline aqueous solution at ambient temperature conditions2.
Why Capability Computing using JUQUEEN
First of all, β-elimination reactions involve atoms that are not directly located at the disulfide bond, which requires a rather large molecular model to study. Secondly, the solvent is critically involved in the process which imposes to use explict water molecules with full electronic structure in a periodic supercell setup. Next, it is required to sample multi-dimensional free energy surfaces at finite temperatures in order to investigate the different reaction pathways. Last but not least, these ab inito metadynamics simulations need to be carried out at several forces in order to find out if reaction scenarios change at specific critical forces. Overall, it is required to simulate a chemically reactive solution composed of about 400 atoms in total, treated at the electronic structure level using density functional theory, in order to sample its statistical mechanics at the level of reactive free energy landscapes using ab initio metadynamics3. In view of the excellent scaling properties of the CPMD program package4, the Blue Gene/Q system at Jülich Supercomputing Centre, JUQUEEN, turned out to be an ideal platform to carry out this demanding project.
From the Expected Carbanion Reaction to a Carbocation Pathway
We reveal that the rate–determining first step of the thermal reaction, which is abstraction of the β-hydrogen, is insensitive to external forces. However, forces larger than about 1 nN are found to reshape the free energy landscape of the reaction so dramatically that a second channel is created, where the order of the reaction steps is reversed – turning β-hydrogen abstraction into a barrierless follow–up process to C–S cleavage. This transforms a slow and force–independent second–order kinetics into a unimolecular reaction that is greatly accelerated by mechanical forces2.
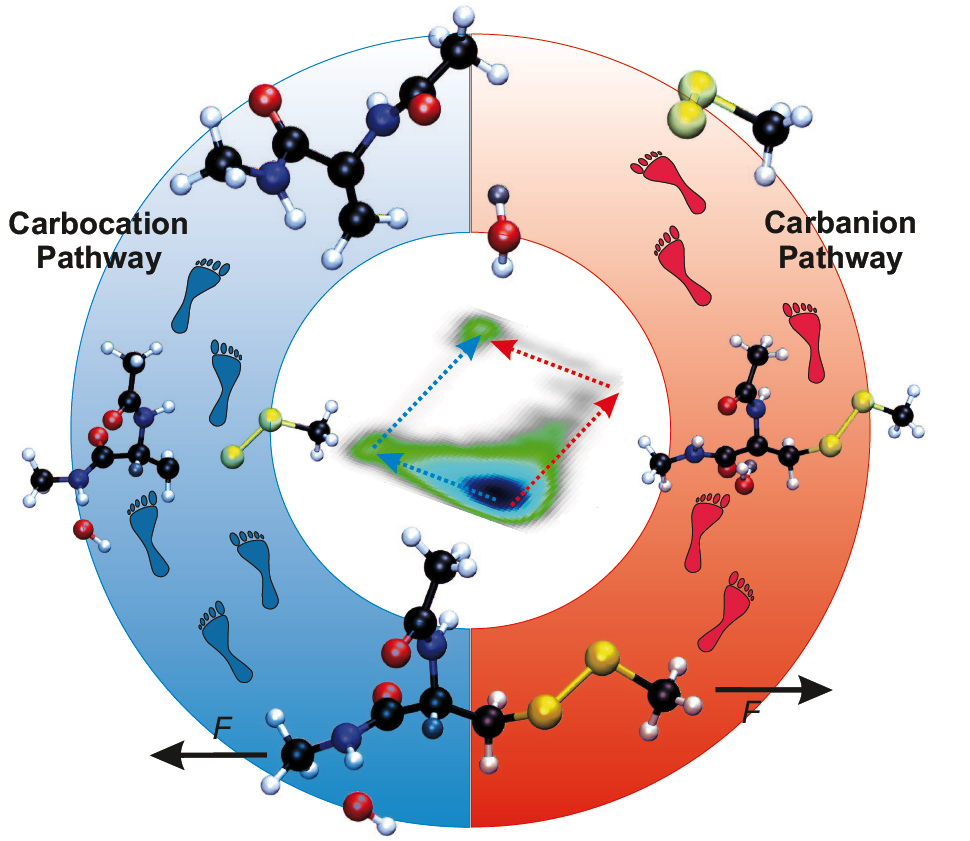
In particular, in the absence of external forces, i.e. in the so-called thermal limit, and still at sufficiently small forces a carbanion intermediate is formed in the first step, which is rate-limiting based on its activation free energy. The second step, which is pure C–S bond rupture, is an almost barrierless and thus a spontaneous follow-up reaction. In stark contrast, at intermediate and high forces, about 1 nN and beyond, pure C–S bond rupture is found to be the first step, thus forming a carbocation, followed by the abstract of the β–hydrogen by OH− which yields a water molecule. The low-force carbanion and high-force carbocation pathways are illustrated in the figure in terms of structural snapshots.
At the level of detailed mechanisms, it is disclosed that a so-called hydrogen–bonded intermediate state (HBIS) is formed which stabilizes the reactant complex. This implies that describing the desolvation and resolvation processes around attacking agent in terms of hydrogen-bond changes and proton transfer reactions is of paramount importance to properly study these complex mechanochemical reactions in aqueous solutions.
Acknowledgements:
This project has been partially funded by the Deutsche Forschungsgemeinsschaft in terms of the Reinhart Koselleck project “Understanding Mechanochemistry” (MA 1547/9) of D.M. and is also part of the Cluster of Excellence “RESOLV” (EXC 1069).
References:
1. J. Ribas-Arino and D. Marx, Covalent mechanochemistry: theoretical concepts and computational tools with applications to molecular nanomechanics, Chem. Rev., 112, no. 10, 5412–5487, 2012.
2. P. Dopieralski, J. Ribas-Arino, P. Anjukandi, M. Krupicka, and D. Marx, Force–induced reversal of β–eliminations: stressed disulfide bonds in alkaline solution, Angew. Chem. Int. Ed., 55, 1304–1308, 2016.
3. D. Marx and J. Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, Cambridge, 2009.
4. J. Hutter and et al., CPMD Program Package.
Scientific team and contact:
Przemyslaw Dopieralski1,2, Jordi Ribas–Arino1,3, and Dominik Marx1 (PI)
1 Lehrstuhl für Theoretische Chemie, Ruhr–Universität Bochum, 44780 Bochum, Germany
e-mail: {przemyslaw.dopieralski, jordi.ribas, domink.marx}@theochem.rub.de
2 Present Address: Faculty of Chemistry, University of Wroclaw, Joliot–Curie 14, 50-383
Wroclaw, Poland
e-mail: mclar@elrond.chem.uni.wroc.pl
3 Present Address: Departament de Qu´ımica F´ısica and IQTCUB, Universitat de Barcelona, Av. Diagonal 647, 08028, Barcelona, Spain
e-mail: jribasjr@yahoo.es
November 2016
JSC Project ID: hbo38