
MATERIALS SCIENCE AND CHEMISTRY
First Principles Multiscale Kinetic Modelling of Catalytic Reactions
Principal Investigator:
Karsten Reuter
Affiliation:
Lehrstuhl für Theoretische Chemie, Technische Universität München (Germany)
Local Project ID:
pr94sa
HPC Platform used:
SuperMUC (LRZ)
Date published:
Researchers at the Technical University of Munich study surface catalytic processes at a variety of scales, combining several different theoretical methods. They take into account the molecular scale of chemical reactions by first principles calculations of thermodynamic adsorption energies and kinetic reaction barriers. These calculations serve as input for mesoscopic models, which include the statistical interplay between the various chemical reactions and allow to predict macroscopic reaction rates and product selectivities. The work provides new insight into the mechanisms of catalytic reactions and gives important leads how to design improved catalyst materials.
Introduction
In later years, the growing demands for alternative power sources and rising concerns about global warming has renewed the interest in developing efficient processes for the synthesis of chemicals and fuels using solid (heterogeneous) catalysts. A mixture of carbon monoxide and hydrogen (also known as syngas) is the starting point for many reactions of technological interest. The obtained end products are highly dependent on the solid material that catalyzes the reaction. Often the catalytically active material is present in the form of nanoscopic particles of transition metals (e.g. Co, Ru, Rh, Ni, Cu or alloys) distributed on a porous support material. In order to tune the activity of the catalyst and the selectivity towards the desired end product (e.g. methane, longer chain hydrocarbons, methanol or ethanol) it is important to gain an atomic-scale understanding of the working catalyst. At the Technical University of Munich (TUM) scientists study catalytic processes at surfaces with a multiscale modelling approach, ranging from the microscopic elementary processes taking place at the catalytic surface to the construction of a microkinetic model that takes into account the statistical interplay between those processes and allows to predict the macroscopic reaction rate and product selectivity. This theoretical modelling relies on an extensive database of first principles density functional theory (DFT) calculations, the construction of which requires the use of high performance computing facilities such as LRZ’s SuperMUC.
The Challenge
The complexity of catalytic reactions on surfaces calls for efficient means of estimating both thermodynamic adsorption energies of the different species involved in the process and kinetic reaction barriers for the considered reaction steps, which are required input for a microkinetic model. For predictive-quality input data DFT is generally the method of choice owing to its favorable ratio between accuracy and computational cost. Still, the large system sizes required for a faithful model of real catalysts, along with the large number of calculations that need to be performed for complex reactions and the consideration of several (or the screening of many) different catalyst materials, renders this approach very challenging. A commonly used strategy to simplify this challenge is to employ scaling relations, which are linear relations between the adsorption energies of chemically related species. Of particular high computational cost is the calculation of reaction barriers (equivalently known as activation energies), which involves identification of the transition state of the reaction step. This is illustrated for the dissociation of a water molecule in Figure 1(a). In Figure 1(b) is shown an example of a scaling relation, i.e. the activation energy of the reaction step scales linearly with the reaction energy. The latter quantity involves only thermodynamic adsorption energies, which are computationally much less costly to obtain than kinetic barriers. Scaling relations are therefore useful for predicting kinetic barriers on new materials (e.g. TM alloys) at a much lower computational cost than the explicit DFT calculation of all reaction energetics.
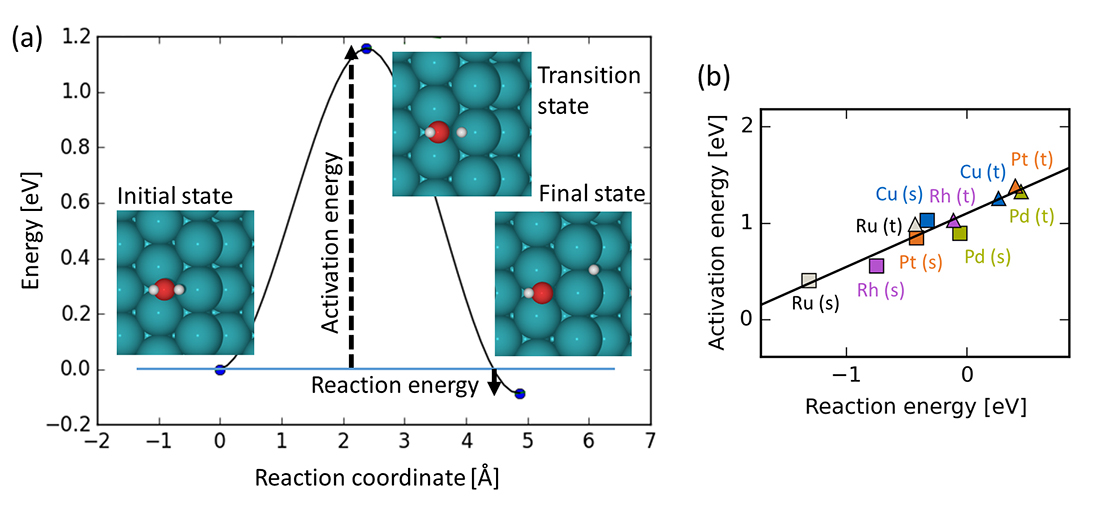
Figure 1: (a) DFT-calculated energies and structures of the initial, transition and final state for the dissociation of a water molecule on a terrace site of a Rh(211) catalyst. (b) Scaling relation between the activation energy and the reaction energy of the water dissociation step illustrated for terrace (t) and step (s) sites on the (211) facet of several transition metals. Based on data from Ref. [1].
Copyright: Technische Universität MünchenResults
The work carried out at TUM addresses several different levels of theory involved in the multiscale modelling of catalytic reactions (c.f. Figure 2). A fundamental and critical choice for the theoretical modeling is the type of active site(s) to focus the investigations on [2]. For many reactions in heterogeneous catalysis this could be a specific site type on a TM nanoparticle such as a surface step or terrace site [1], but it could equivalently be a site at the interface between a TM and an oxide material [3]. Next, the elementary processes that can take place on the active site need to be considered for the particular reaction of interest. As discussed above, the reaction energetics is typically calculated from DFT, while scaling relations can be used to extend the theoretical predictions to many similar materials such as the TM and their alloys [1]. This is typically the part of the multiscale modelling approach that relies most heavily on supercomputing power.
The DFT-calculated reaction energetics allows to calculate rate constants for all elementary processes, which serve as input to a microkinetic model. For the prediction of rough trends and limitations in catalyst activities, the microkinetic model often relies on a mean-field approximation (MFA), in which a random spatial distribution of the chemical species on the catalyst surface is assumed. This approach has been used to estimate upper limits to the catalytic performance of bifunctional catalysts [4-5], which are special type of catalysts that rely on the coupling of two different active sites, each catalyzing a particular reaction step. Such insights serve to provide ideas how to design new catalyst materials that go beyond the limitations to the optimal catalyst activity achievable posed by scaling relations.
To go beyond the MFA and explicitly account for spatial correlations in the distribution of the chemical species on the surface, scientist at TUM make use of kinetic Monte Carlo (kMC) simulations. For the specific reaction of carbon monoxide and hydrogen to form methane, explicit assessment of the MFA against the more elaborate kMC simulations showed that the MFA can break down for step sites on metal surfaces. This was shown to arise from the fact that these sites bind the chemical species very strongly and therefore hinder their mixing on the catalyst surface [1]. The work underlines the importance of considering more advanced microkinetic models despite their higher computational cost, also for the screening of new catalyst materials where the MFA has traditionally been applied.
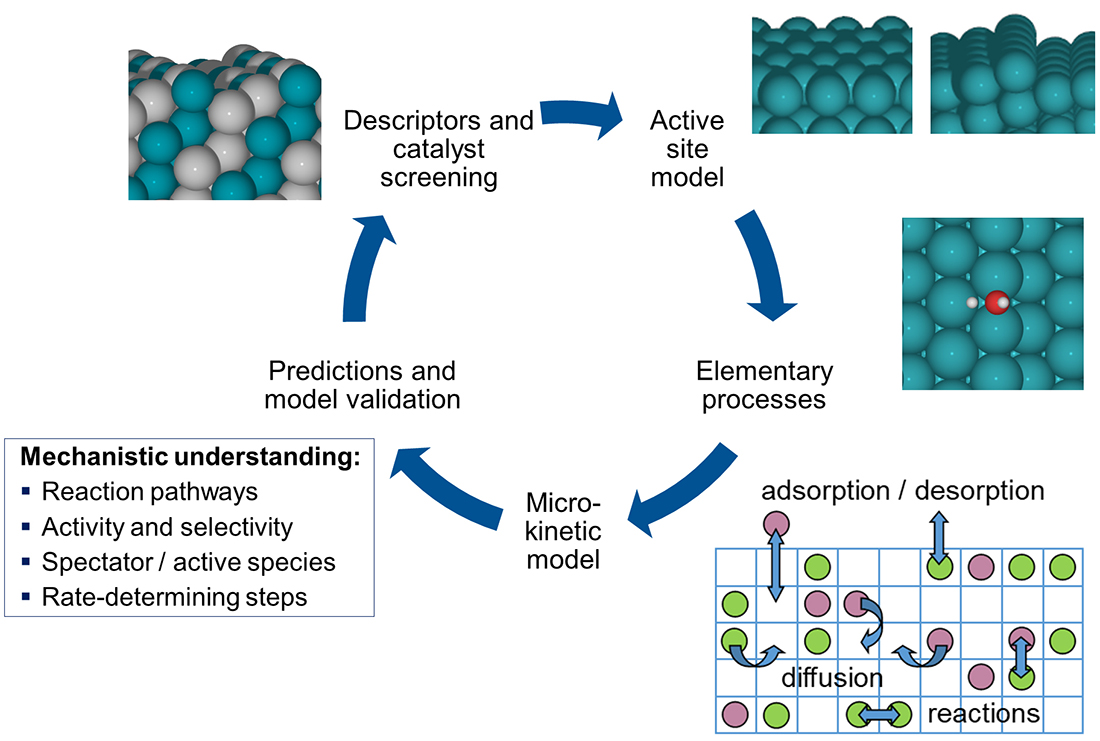
Figure 2: Overview of the various levels of theory involved in the multiscale modelling of catalytic reactions. The reaction energetics of the elementary processes is typically calculated at one or more active site models using DFT. This serves as input to a microkinetic model, which is typically constructed using either the mean-field approximation or kinetic Monte Carlo simulations. From the microkinetic model a number of predictions can be made related to e.g. the dominant reaction pathway as well as to which reaction steps within that pathway that are most important for the observed catalytic activity and product selectivity. Finally, the obtained insight can be used to identify descriptors for catalytic performance, which in turn facilitate the search for new and improved catalyst materials through computational catalyst screening.
Copyright: Technische Universität MünchenAcknowledgments:
Mie Andersen would like to thank the Alexander von Humboldt Foundation for a post-doctoral fellowship. Funding through the German Research Foundation (DFG) is acknowledged within Project No. RE1509/27-1.
References:
[1] M. Andersen, C. Plaisance, and K. Reuter, J. Chem. Phys. 147, 152705 (2017)
[2] K. Reuter, C. Plaisance, H. Oberhofer, and M. Andersen, J. Chem. Phys. 146, 040901 (2017)
[3] M. Andersen, X. Yu, M. Kick, Y. Wang, C. Wöll, and K. Reuter, J. Phys. Chem. C 122, 4963 (2018)
[4] M. Andersen, A.J. Medford, J.K. Nørskov, and K. Reuter, Ang. Chem. Int. Ed. 55, 5210 (2016)
[5] M. Andersen, A.J. Medford, J.K. Nørskov, and K. Reuter, ACS Catal. 7, 3960 (2017)
Scientific Contact:
Dr. Mie Andersen
Theoretische Chemie, Technische Universität München
Lichtenbergstraße 4, D-85747 Garching/Munich (Germany)
E-mail: mie.andersen [at] ch.tum.de
http://www.th4.ch.tum.de/andersen