
MATERIALS SCIENCE AND CHEMISTRY
Theoretical Heterogeneous Catalysis from Advanced Ab Initio Molecular Dynamics Simulations
Principal Investigator:
Dominik Marx
Affiliation:
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, Bochum (Germany)
Local Project ID:
pr63ce
HPC Platform used:
SuperMUC (LRZ)
Date published:
The work horse of chemical industry is heterogeneous catalysis meaning that complex solid materials (catalysts) are used to facilitate chemical reactions, thus reducing production costs. To improve such catalysts in a systematic manner, knowledge of the ongoing reactions is most desirable. One of the key reactions industry performs at large scales is methanol (“wood alcohol”, H3COH) synthesis from syngas being a mixture of gaseous CO2, CO, and H2. Scientists investigated the methanol production which is catalyzed using copper nanoparticles on a zinc oxide support. Based on sophisticated molecular dynamics sampling techniques in conjunction with the large-scale parallel platform SuperMUC at LRZ, they discovered a hitherto unknown complex reaction network with many parallel paths as well as dead ends that ultimately leads to the reaction of syngas to methanol.
Introduction
The work horse of chemical industry is heterogeneous catalysis meaning that complex solid materials (catalysts) are used to facilitate chemical reactions, thus reducing production costs. To improve such catalysts in a systematic manner, knowledge of the ongoing reactions is most desirable. One of the key reactions industry performs at large scales is methanol (“wood alcohol”, H3COH) synthesis from syngas being a mixture of gaseous CO2, CO, and H2. Scientists investigated the methanol production which is catalyzed using copper nanoparticles on a zinc oxide support. Based on sophisticated molecular dynamics sampling techniques in conjunction with the large-scale parallel platform SuperMUC at LRZ, they discovered a hitherto unknown complex reaction network with many parallel paths as well as dead ends that ultimately leads to the reaction of syngas to methanol.
The Challenge
The synthesis of methanol is a large-scale process in chemical industry. Nowadays methanol is produced from syngas (a mixture of CO2, CO, and H2) over the Cu/ZnO catalyst at 50-100 bar and 250°C, which is the so-called “ICI process”. At these rather harsh conditions, the catalyst undergoes significant dynamical changes of its shape and chemical properties. These changes allow for a manifold of different positions where the participating molecules of the reaction can absorb and thus become catalytically activated to undergo chemical reactions. This implies that a single catalyst may facilitate several reactions depending on the actual chemical properties of the adsorption site. The situation is even more complicated since the shape and the chemical properties of the catalyst itself depend on the molecular composition as well as the thermodynamic state (determined by temperature and pressure) of the surrounding gas phase [1,2].
Due to the complexity of the reaction network of most heterogeneously catalyzed processes the rational design of new and improved catalysts is a daunting task. Especially the quite harsh reaction conditions hamper obtaining insights of ongoing reactions via experiments. Scientists use computer simulations to overcome this limitation aiming at a detailed molecular picture of the chemical processes as a step toward rational catalyst design.
Necessity for HPC Facility
First of all, simulations of surfaces require a model system containing a rather large number of atoms. The model has to capture properties from an extended solid and the interaction with a gas phase. In the case of a supported catalyst as Cu/ZnO, the surface has to be large enough to host the supported material and to expose free surface to the gas phase.
Simulations of dynamical properties require molecular dynamics techniques, where the movement of individual atoms is calculated. To allow for chemical reactions, the electrons have to be included explicitly, requiring ab initio molecular dynamics techniques [3]. The drawback of ab initio techniques is the huge computational cost, even if highly efficient state-of-the-art simulation packages as CPMD [4] in the present case are used. Another aspect increasing the computational effort is the statistics needed to estimate the reaction network and to identify each involved reaction. This is done via calculation of a free energy landscape or surface (FES) along certain degrees of freedom of interest. Reactions are paths with lowest free energy connecting two valleys via a pass. The FES can be sampled using the so-called multiple walker technique to increase the efficiency that lends itself to parallel processing, yet required powerful individual nodes. The SuperMUC system turned out to be the ideal platform to carry out these simulations.
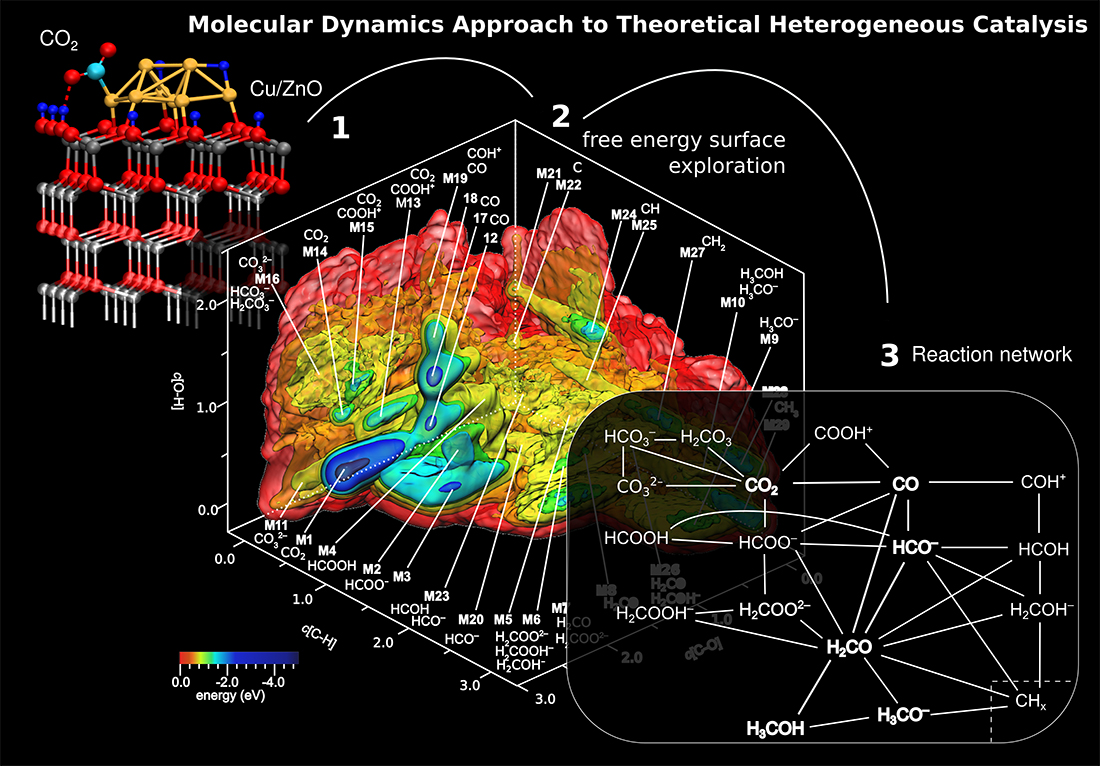
Figure 1: Process of generating a global reaction network of chemical species and elementary reactions from pure computational means. (1) Thermodynamic optimization: Activated CO2over the Cu8/ZnO catalyst model. (2) Metadynamics sampling: 3D free energy surface with the “synthesized” chemical species which were obtained from analysis of the ab initio molecular dynamics trajectory underlying the metadynamics. (3) Schematic reaction network of methanol synthesis from CO2 over the Cu8/ZnO nanocatalyst model including parallel reactions, prominent side reactions such as methanation, reverse and forward water gas shift reaction, and reactions leading to catalyst deactivation. Picture adapted from Ref. [5].
Copyright: Ruhr-Universität BochumResults
The molecular dynamics approach to theoretical heterogeneous catalysis [6-8] was applied to the methanol synthesis over Cu/ZnO catalyst. The model system was carefully optimized for the temperature and pressure of the industrial process [1,2]. The simulated 3D free energy landscape shows the chemical transformation of molecular carbon species from CO2 towards methanol, see Figure 1 and Ref. [5]. Analysis of the complex landscape discloses an overwhelmingly rich reaction network of parallel, competing, and reverse reactions over the lively Cu/ZnO catalyst surface. More than twenty molecular carbon species have been generated in ab initio metadynamics, see Figure 1 herein.
The key route from CO2 towards methanol is the reaction via formaldehyde (H2CO), which is a highly reactive intermediate species. In addition, the analysis revealed that all known side reactions (reverse and forward water gas shift, methanation) have been generated including reactions deactivating the catalyst such as coaking. Further analysis identified three prototypical surface reaction mechanism discussed among scientists, namely the so-called Eley-Rideal, Langmuir-Hinshelwoold, and Mars-van Krevelen types. The former and the latter mechanism can only be realized if the simulation explicitly considered all the relevant parts, namely the near surface at the interface between catalyst and gas phase. As example, Figure 2 shows the methanol synthesis from formaldehyde via the Eley-Rideal mechanism. In addition, it illustrates the dynamical transformation of the catalyst.
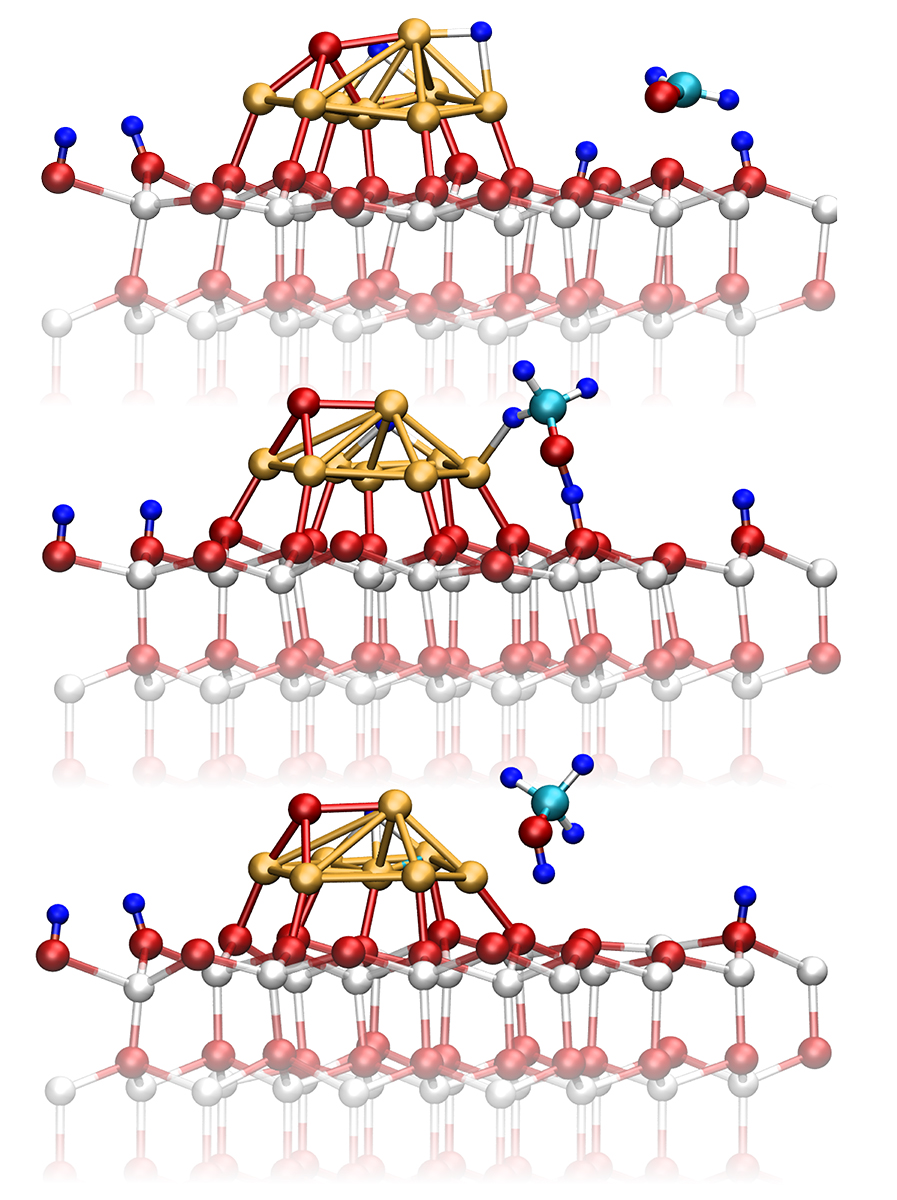
Figure 2: Potential reaction pathway of methanol formation from formaldehyde over the Cu8/ZnO nanocatalyst model via the Eley–Rideal type surface chemical reaction. Representative snapshots are taken from the ab initio molecular dynamics trajectory underlying the sampling of the free energy surface as shown in Figure 1. Picture taken from Ref. [5].
Copyright: Ruhr-Universität BochumOutlook
The presented detailed knowledge of the reaction mechanisms and the reaction network of industrial methanol synthesis according to the ICI process may help industrial chemists in their future research efforts to improve this catalyst. Our own future work will focus on disclosing the mechanism of selective alcohol oxidation using gold-titania nanocatalysts with a particular focus on liquid-phase heterogeneous catalysis.
Acknowledgements
This work has been supported by the German Research Foundation (DFG) via the Collaborative Research Center SFB 558 “Metal–Substrate Interactions in Heterogeneous Catalysis” and the Cluster of Excellence EXC 1069 “RESOLV”.
References:
[1] L. Martínez-Suárez, J. Frenzel, D. Marx, and B. Meyer, Phys. Rev. Lett. 110, 086108 (2013).
[2] L. Martínez-Suárez, J. Frenzel, D. Marx, Phys. Chem. Chem. Phys. 16, 26119 (2014).
[3] D. Marx and J. Hutter, “Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods”, Cambridge University Press (2009).
[4] CPMD program package: http://www.cpmd.org/.
[5] L. Martínez-Suárez, N.Siemer, J. Frenzel, D. Marx, ACS Catal. 5, 4201 (2015).
[6] J. Kiss, J. Frenzel, N. N. Nair, B. Meyer, and D. Marx, J. Chem. Phys. 134, 064710 (2011).
[7] J. Frenzel, J. Kiss, N. N. Nair, B. Meyer, and D. Marx, Phys. Status Solidi B 250, 1174 (2013).
[8] J. Frenzel, D. Marx, J. Chem. Phys. 2 141, 124710 (2014).
Scientific Team:
Prof. Dr. Dominik Marx (PI), Dr. Johannes Frenzel, Dr. Daniel Muñoz-Santiburcio, Dr. Luis Martínez-Suárez, M. Sc. Niklas Siemer
Scientific Contact:
Prof. Dr. Dominik Marx
Lehrstuhl für Theoretische Chemie - Ruhr-Universität Bochum
Universitätsstraße 150, D-44801 Bochum (Germany)
e-mail: dominik.marx[at]rub.de