
MATERIALS SCIENCE AND CHEMISTRY
Mechanochemistry or the “Strange case of Dr. Jekyll and Mr. Hyde”
Principal Investigator:
Dominik Marx
Affiliation:
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum
Local Project ID:
hbo38
HPC Platform used:
JUQUEEN of JSC
Date published:
In order to initiate chemical reactions, energy must be provided in order to overcome the so-called activation barrier, which separates the reactants from the product of the reaction. This energy can be supplied in different forms such as heat, light, electrical current or as mechanical forces that distort the molecules involved.
In order to achieve that experimentally, two long polymer chains are attached to the reactive molecule, which is called “mechanophore” in analogy to “chromophores” widely used in photochemistry. These chains serve as ropes to stretch out the mechanophore, either by pulling at them in a single-molecule AFM setup, or as a result of sonicating a solution of these engineered molecules. The more mechanical energy is put into the molecule, meaning: the harder it is pulled, the more reactive it usually is. But this is not always the case!
Recent, most enigmatic experiments in this field come from studying the reactivity of disulfide bridges embedded in proteins once they get stretched in an AFM setup at constant force conditions. In particular, two different reactivity regimes have been observed. At small external forces the activation of the reaction has been found to be very pronounced, whereas its acceleration becomes much less efficient beyond some critical force. Although several possible explanations have been proposed to explain this puzzling result in various publications, all of them remained inconclusive or even contradictive.
Mechanochemistry or the “Strange case of Dr. Jekyll and Mr. Hyde”Copyright: © Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum
Why High Performance Computing using JUQUEEN
To tackle this problem novel mechanochemical simulation techniques (such as isotensional ab initio metadynamics and thermodynamic integration) in conjunction with the power of the Blue Gene/Q platform JUQUEEN at Jülich Supercomputing Centre (JSC) have been used.
In all previous calculations significant approximations had been made, such as using continuum or microsolvation models, carrying out the calculations in the absence of thermal fluctuations (i.e. at 0 K) or strongly biasing reaction pathways. All this obviously reduces the computational cost, but at the same time reduces the predictive power of such studies.
In the novel approach, however, it is not necessary to resort to such drastic simplifications, thus the reactive core, i.e. the disulfide bridge connected to the backbone, has been treated by a fully solvated diethyl disulfide molecule (see Figure), which is attacked by OH− hosted in the same periodic simulation cell.
Furthermore, using ab initio metadynamics, scientists were able to map the free energy surface in a rather general reactive subspace where the reaction coordinate can be expressed. Such simulations are already very demanding by themselves, which is tackled by using parallelized multiple walker sampling (as implemented by Marx group in the CPMD code for Blue Gene platforms in the frame of another project: N. N. Nair, E. Schreiner, and D. Marx in inSiDE (Innovatives Supercomputing in Deutschland) 6(2), 30–35 (2008): “Prebiotic Peptide Synthesis on Blue Gene Platforms at ‘Iron–Sulfur–World’ Conditions”). In the present case, however, such simulations need to be carried out isotensionally, i.e. as a function of external force, and require, in addition, an unusually high statistical quality in order to pinpoint the critical force.
In summary, the key to success was a particularly complex version of molecular simulation in conjunction with computer time allocation in the frame of a GCS Large–Scale Project.
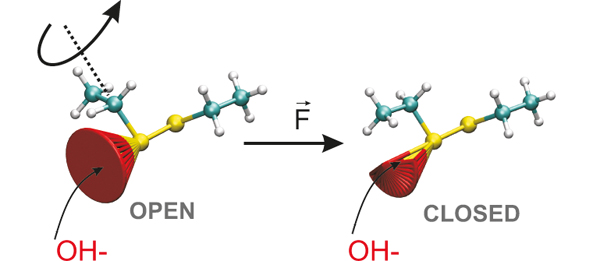
Simplified sketch of the change in the reaction cone (red) of the observed reaction, i.e. bond cleavage as a result of nucleophilic attack by OH− in bulk aqueous solution. The reaction cone closes as a result of mechanical pulling (right panel) due to applying an external force F to the terminal carbon atoms, which sterically hinders the reaction at sufficiently large forces. The S atoms of the disulfur bridge are represented by yellow spheres, whereas the C and H atoms of the two ethyl linker groups are shown in grey and turquoise. © Lehrstuhl für Theoretische Chemie, Ruhr–Universität Bochum
Fundamental Insights into Mechanochemical Processes
First of all, performed complex simulation was the pioneering one to reproduce the experimentally observed peculiar reactivity switch in essentially quantitative agreement. This made the researchers confident to analyze the underlying mechanism, which is pictorially explained in the Figure. The key finding is that applying force not only straightforwardly lowers reaction barriers, as widely believed earlier, but also distorts the stretched molecule in such a way that the attacking OH− cannot reach the reaction center as easily as in the unstrained molecule at low forces. The full results have been published by Dopieralski et al. in Nature Chemistry 5, 685–691 (2013): “The Janus-faced role of external forces in mechanochemical disulfide bond cleavage”. Beyond the specific case, presented mechanistic discovery might well prove to be most valuable in mechanochemistry as such since, in general, molecular distortion is expected as a result of tensile stress. Similar ideas might hold as well in the regulation of biomolecular function in organisms, which is a topic of future research.
This project has been funded by the Deutsche Forschungsgemeinsschaft in terms of the Reinhart Koselleck project “Understanding Mechanochemistry” of D.M.
Przemyslaw Dopieralski1,2 , Jordi Ribas–Ariño3, and Dominik Marx1
1 Lehrstuhl für Theoretische Chemie, Ruhr–Universität Bochum, 44780 Bochum, Germany
e-mail: {przemyslaw.dopieralski, domink.marx}@theochem.rub.de
2 Faculty of Chemistry, University of Wroclaw, Joliot–Curie 14, 50-383 Wroclaw, Poland
e-mail: mclar@elrond.chem.uni.wroc.pl
3 Departament de Química Fisica and IQTCUB, Universitat de Barcelona, Av. Diagonal 647, 08028, Barcelona, Spain
e-mail: jribasjr@yahoo.es