
MATERIALS SCIENCE AND CHEMISTRY
Studying Structure to Understand Function in Phases of the Elements
Principal Investigator:
Robert O. Jones
Affiliation:
Forschungszentrum Jülich (Germany)
Local Project ID:
hpg00
HPC Platform used:
JUQUEEN of JSC
Date published:
“If you want to understand function, study structure” (F. Crick). In the case of carbon, the very different properties of graphite, diamond, and carbon nanotubes can then be traced to different atomic arrangements. The elements provide a fruitful field of study in general. There are fewer than 100 stable elements, and trends can be identified more readily than in alloys with infinitely many compositions. He we describe cluster, amorphous, and liquid phases of the group 15 elements bismuth and antimony using molecular dynamics simulations based on density functional theory.
The molecular biologist and geneticist Francis Crick wrote: “If you want to understand function, study structure” [1]. An example is provided by forms of carbon, where diamond, graphite, and fullerene cages, not to mention graphene and carbon nanotubes, differ only in their atomic arrangements, yet have quite distinct properties. There are also intriguing differences between the properties of related elements, an example being the group 14 elements (valence electron configurations s2p2) with electronic properties ranging from insulators (C) to semiconductors (Si and Ge), and to metals (Sn and Pb).
Can one understand these and other trends within families of elements? There are relatively few—less than 100—stable elements, so that trends are often easier to discern than in alloys of several elements with infinitely many possible compositions. Our group has studied such trends for many years, with particular interest in group 16 elements (S, Se, Te). The present project has focused on the group 15 elements antimony and bismuth, particularly the cluster and liquid phases. A particular challenge is provided by the crystallization of supercooled liquid Sb, which is known to occur spontaneously and explosively.
These questions can be addressed by molecular dynamics (MD) simulations on the basis of density functional (DF) theory [2]. DF calculations are free of adjustable parameters and have given valuable predictions in many contexts, but DF/MD calculations are numerically very demanding. The time step in the simulations is very short, and even very rapid processes in disordered materials require many calculations of forces and energies for large samples (hundreds of atoms). Supercomputers are essential for this task, and the work at the FZ Jülich has used the GCS supercomputer JUQUEEN.
A decisive motivation for the project was the availability of high-quality inelastic x-ray scattering (IXS) and neutron diffraction data. The position and velocity of each atom are stored at each step of an MD simulation, and it is possible to compare dynamical information (scattering factors, viscosities, sound velocities) with measurements. Moreover, atomic positions can be inferred only indirectly from scattering measurements, so that theory and experiment are truly complementary.
The structures of Bi and Sb clusters with up to 14 atoms [3,4] follow trends familiar in the lighter group 15 elements P and As, but the very extensive simulations of liquid Bi [3,5] and Sb [4,6] provided much new information. A very pleasing result in Bi was the excellent agreement with recent IXS measurements of the dynamical structure factor. Alfred Baron, one of the experimentalists involved, noted that the agreement between theory and experiment ‘is really quite beautiful,’ so that even small differences could provide valuable information. The quality of agreement between theory and experiment was unexpected, but confirmed that simulations and experiments are complementary and that the level of agreement can be astonishingly good—even for ‘real’ materials. However, it also showed that extensive and time-consuming simulations are essential if detailed agreement is to be achieved.
The simulations of liquid Sb at eight temperatures between 1300 K and 600 K (supercooled) provided very interesting results, and IXS measurements are now being carried out. Spontaneous crystallization was not observed, however, even after 570 ps. Crystallization occurred readily in the presence of crystalline templates of various forms, as shown in Fig. 1, where crystallization took less than 50 ps in the presence of a crystalline substrate. We have not been able to resist the temptation to move on to other elements, and we are focusing now on the structure of amorphous Se, one of the best studied materials of all.
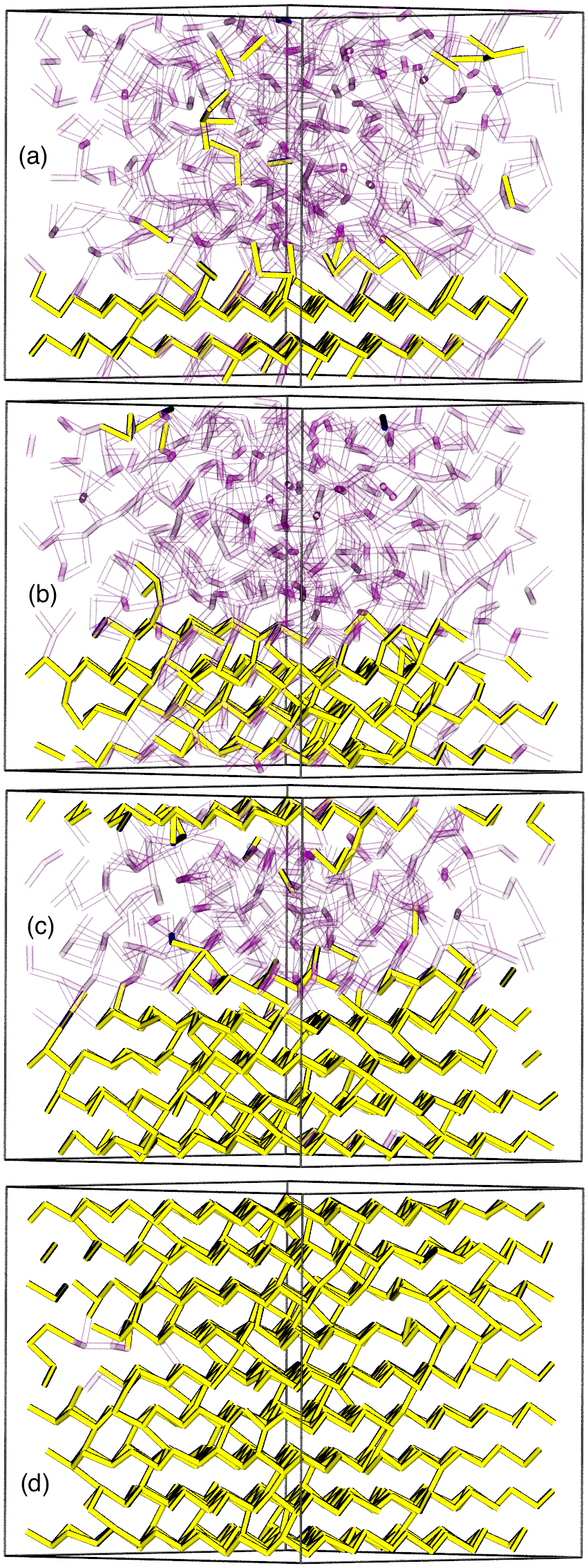
Crystallization of 882-atom simulation of Sb in the presence of a crystalline substrate. After (a) 0 ps, (b) 13 ps, (c) 30 ps, (d) 46 ps. Yellow tubes connect crystalline atoms, transparent purple tubes disordered atoms.
Copyright: FZ JülichPublications:
[1] F. Crick: What Mad Pursuit (Penguin, London, 1990), p 150
[2] R. O. Jones: Density functional theory: Its origins, rise to prominence, and future
[Online-Edition: http://doi.org/10.1103/RevModPhys.87.897]
In: Rev. Mod. Phys. 87, 897 (2015)
[3] J. Akola, N. Atodiresei, J. Kalikka, J. Larrucea, and R. O. Jones: Structure and dynamics in liquid bismuth and Bin clusters: A density functional study [Online-Edition: http://doi.org/10.1063/1.4901525]
In: J. Chem. Phys. 141, 194503 (2014)
[4] R. O. Jones, O. Ahlstedt, J. Akola, and M. Ropo: Density functional study of structure and dynamics in liquid antimony and Sbn clusters
[Online-Edition: http://doi.org/10.1063/1.4983219]
In: J. Chem. Phys. 146, 194502 (2017)
[5] M. Ropo, J. Akola, and R. O. Jones: Collective excitations and viscosity in liquid Bi
[Online-Edition: http://doi.org/10.1063/1.4965429]
In: J. Chem. Phys. 145, 184502 (2016)
[6] M. Ropo, J. Akola, and R. O. Jones: Crystallization of supercooled liquid antimony: A density functional study
[Online-Edition: http://doi.org/10.1103/PhysRevB.96.184102]
In: Phys. Rev. B 96, 184102 (2017)
Research Team:
Jaakko Akola (Norwegian University of Science and Technology, Trondheim, Norway), Janne Kalikka, Matti Ropo (Tampere University of Technology, Finland)
Scientific Contact:
Robert O. Jones
Forschungszentrum Jülich
Peter Grünberg Institut PGI-1, D-52425 Jülich (Germany)
e-mail: r.jones [@] fz-juelich.de
July 2018
JSC project ID: hpg00