MATERIALS SCIENCE AND CHEMISTRY
Effects of Lattice Dynamics on the Absorption of Strongly Anharmonic Semiconductors
Principal Investigator:
Prof. David Egger
Affiliation:
TUM School of Natural Sciences, Technical University of Munich, Germany
Local Project ID:
lattdynabs
HPC Platform used:
JUWELS CPU at JSC
Date published:
Abstract
Halide perovskites are booming as absorber materials for solar cells: they are cheap and easy to make in the lab while delivering devices with efficiencies of converting the energy of sunlight into electricity. An interesting, more fundamental aspect of these materials regards their atomic motions, which are unusual compared to other solar materials. Researchers at the Technical University of Munich used the power of the JUWELS cluster to investigate the impact of these atomic motions on the absorption of sunlight in halide perovskites. Their findings were intriguing: the strong atomic motions do not hinder but are in actuality a beneficial feature for the efficient collection of sunlight in halide perovskites.
Results and Methods
Limiting the increase of the average earth’s temperature to below 2 degrees Celsius requires the reduction of green-house gas emissions. At the same time, we are confronted with the challenge of supplying enough energy to the constantly growing demands of human societies. A promising solution to these two issues is development and deployment of novel renewable energy technologies. Photovoltaics is the generation of electricity from capturing sunlight in solar cells, which probably marks one of the most important and popular technologies in this context. We can see this by witnessing the growing amount of solar panels all around us.
For decades, researchers have focused their efforts on developing new materials that can absorb sunlight, and turn it into electricity, in an ever more efficient way. Among the most promising, recent materials for solar cells are halide perovskites. Perovskites are long-known as minerals occurring in nature. Usually containing oxygen, the halide versions of these substances instead feature ions from the halogen series, which are found in the penultimate column of the periodic table. These halide perovskites do not occur in nature but are synthesized in the lab. A key aspect of these materials is precisely that they are so easy to synthesize, yet that they also deliver semiconductors for solar cells with remarkable optical and electrical properties. At the same time, one can mix different kinds of ions – metal and halogen species – to fine-tune the material properties and tailor them to a specific technological application.
Halide perovskites not only exhibit favorable synthesis and optoelectronic properties, they also show very peculiar motions of their constituent atoms: large displacements occur in these crystals with a thermal behavior that is very different from that observed in more conventional semiconductor crystals, like Silicon – the material in most solar panels we see around us, or Gallium Arsenide which is another well-known solar material. The particular atomic dynamics in the halide perovskite do not follow the typical, harmonic and wave-like appearance that are known in these “classical” semiconductors. Instead, the atomic motions in halide perovskites are anharmonic and at times appear particle- instead of wave-like. This introduces disorder that usually is thought to be bad for the performance of solar-cell materials. And yet, these perovskites exhibit remarkable optoelectronic and efficiency performances, which altogether is very unusual. Understanding how all these properties come together, and detecting the microscopic mechanisms behind them, is an exciting fundamental research question.
To answer this question, molecular dynamics calculations are a powerful theoretical tool because they allow one to simulate how the atoms move under a variety of different physical conditions. What is more, these simulations can be combined with more refined quantum-mechanical calculations of the material’s properties, essentially providing a microscopic view of how the electrons are distributed in energy. While these treatments are computationally heavy, it opens the door to investigate and understand the optoelectronic properties of materials, such as halide perovskites, at finite temperature. Using this approach, researchers at the Technical University of Munich (TUM) investigated the underlying mechanisms of atomic disorder and sunlight absorption in halide perovskites, which is essential for the outstanding performance of halide perovskites in solar cells.
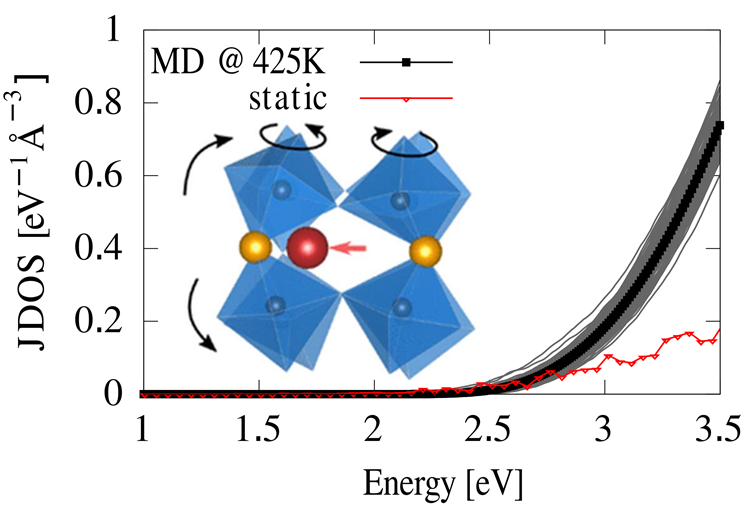
Equipped with the power of the JUWELS supercomputer, the team at TUM was able to simulate the atomic dynamics of large samples of halide perovskite crystals. From the simulated structures, they calculated the frequency of possible electronic transitions involved in sunlight absorption, so-called band-to-band transition profiles (see Figure). By comparing these properties at finite-temperature to those calculated on static, low-temperature structures, the team revealed that anharmonic atomic displacements are beneficial to obtaining sharp optical absorption profiles, an essential feature of efficient solar materials. Since the large atomic motions that were recorded imply a disorder to the crystal structure, these findings challenge the convention according to which a sharp absorption is directly related to very ordered crystals. The team found that this is a peculiar feature of the atomic and electronic structure of halide perovskites, which does not appear in more conventional materials such as Silicon and GaAs. The results of the TUM team open a new perspective on dynamic disorder of the atoms in halide perovskite crystals as a desired feature, which will help the design of new materials with tunable properties, using atomic dynamics as a design metric.
References
- C. Gehrmann, S. Caicedo‐Dávila, X. Zhu, D. A. Egger, Advanced Science. 9, 2200706 (2022).
- X. Zhu, S. Caicedo-Dávila, C. Gehrmann, D. A. Egger, ACS Appl. Mater. Interfaces. 14, 22973–22981 (2022).