
MATERIALS SCIENCE AND CHEMISTRY
Accelerated Materials Discovery with Automation and Machine-Learned Chemical Knowledge
Principal Investigator:
Dr. Janine George
Affiliation:
Friedrich-Schiller-Universität Jena, Institut für Festkörpertheorie und -optik, Jena, Germany
Local Project ID:
pn73da
HPC Platform used:
SuperMUC-NG at LRZ
Date published:
Introduction
This project aims to accelerate the search for new materials (e.g., for thermoelectric applications, battery materials, magnets, and other materials classes) based on ab initio high-throughput studies. High-throughput searches are typically restricted to known materials. This project explores strategies (data-driven chemical heuristics in subproject 1 and machine-learned inter- atomic potentials in subproject 2) to go beyond current database entries and include such computationally demanding properties in high-throughput searches. To accomplish each subproject, we develop automated workflows for high-throughput computations and provide large open databases of computed materials properties to the research community.
Results and Methods
Training data is needed to develop data-driven chemical heuristics and machine-learned interatomic potentials. In computational materials science, we typically rely on density functional theory-based (DFT) training data. Specifically, we use the density-functional theory pack- age VASP [6], very often in combination with the PBE exchange-correlation functional embedded in the PAW method. We typically run these computations on 2-3 nodes with 96-144 CPUs and multiples thereof in the micro or general queue of the cluster.
To compute complex properties such as bonding-based descriptors or vibrational properties of materials, we typically have to combine multiple DFT runs (sometimes up to several hundred) and pre- and post-processing steps (e.g., projection of a plane-wave based basis set on atomic orbitals for bonding properties as implemented in LOBSTER and subsequent analysis of the data with LobsterPy [1,7], or computations of vibrational properties with the finite displacement method as implemented in Phonopy).
Performing high throughput computations of such complex properties would not be possible in a manual way. Therefore, we develop workflow systems, specifically allowing for the demands of computational materials science projects (including options to use different Slurm submission scripts for different projects or jobs, allowing for dynamic generation of new jobs). One of the develop- ments that we are involved in is jobflow [2] - a software for workflow development - and subsequent specific computational materials science workflows as imple- mented in the software atomate2 [3]. All the mentioned developments are open-source projects.
In this way, we have now generated an open quantum-chemical bonding database [4]. We are currently working on the integration of the software in a larger materials properties database that would allow for retrieving the data via an API and, therefore, allow for easier reuse of the data. The workflows and codes to accomplish this are all clearly documented with tutorials.
These bonding properties serve as new descriptors for materials science properties. We then retrieve the relationships between quantum-chemical bonding descriptors and materials properties, making the acceleration of the prediction of materials properties possible in the long run. Based on simple random forest models, we have already shown that the quantum-chemical bonding descriptors indeed have predictive power for vibrational properties [4].
Besides automating DFT runs and pre- and postprocessing steps, machine-learned interatomic potentials can be used within such workflow systems. This allows the automation of their training and benchmarking against DFT data. Our workflow developments for computed vibrational properties with DFT have been extended in such a way that they can now also be used with machine-learned interatomic potentials. We have used this workflow to benchmark a new foundation model for the predtion of phonon properties. While the overall phonon band structures cannot be exactly reproduced, overall bandwidths agree to an acceptable degree, potentially allowing for efficient pre-screening of materials' properties such as (low) thermal conductivity [5].
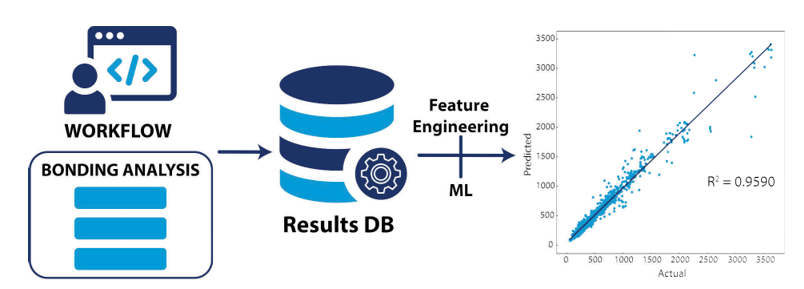
Figure: Schematic representation of the data generation workflow to analysis procedure. Adapted from [4]. Creative Commons CC-BY 4.0.
Ongoing Research/Outlook
Future work will be devoted to providing workflow solutions for the automatic development and benchmark of machine-learned interatomic potentials. This potentially includes efficiently and automatically combining heterogeneous computing resources (CPUs and potentially GPUs).
Furthermore, our dataset of quantum-chemical bonding descriptors is further extended to allow for the development of more deep-learning models, requiring more data.
References and Links
[1] J. George et al., ChemPlusChem 2022, 87, e202200123.
[2] A. S. Rosen et al., J. Open Source Software 2024, 9, 5995.
[3] A. Ganose, et al., github.com/materialsproject/atomate2, 2023.
[4] Naik, A.A., Ertural, C., Dhamrait, N. et al. A Quantum-Chemical Bonding Database for Solid-State Materials. Sci Data 10, 610 (2023). doi.org/10.1038/s41597-023-02477-5
[5] I. Batatiaet al., DOI 10.48550/arXiv.2401.00096.
[6] www.vasp.at