
MATERIALS SCIENCE AND CHEMISTRY
Validating CFD with Large-scale Molecular Dynamics Simulations
Principal Investigator:
Jadran Vrabec
Affiliation:
Chair of Thermodynamics and Process Engineering, Technische Universität Berlin
Local Project ID:
MMHBF2
HPC Platform used:
Hazel Hen and Hawk of HLRS
Date published:
Introduction
Modern engineering cannot be imagined without computational fluid dynamics (CFD). It is omnipresent and used in a wide field ranging from the dimensioning of chemical processes to the development of new airfoils for state-of-the-art airplanes. CFD is based on the solution of the so-called Navier-Stokes equations which describe the behavior of fluids under specific boundary conditions. To conduct CFD simulations reliably, certain terms of the Navier-Stokes equations must be modelled particularly adequately. The modelling accuracy can be assessed e.g. by utilizing direct numerical simulations (DNS). Even though DNS provide good results, this approach has its limits and cannot be applied in specific fields. This is the case e.g. for very small systems or when phase change occurs. At this point, molecular dynamics (MD) comes into play. The underlying principle of the MD is based on the well-known Newton’s law of motion. Each molecule constituting a fluid is considered explicitly, and the aforementioned differential equation is solved for the entire set of the system’s molecules. The only assumption in MD is done for modeling the interactions between the molecules. Hence, MD simulations provide highly accurate results and therefore are particularly suitable for very detailed analyses of a system’s behavior. Since each molecule is considered individually, MD simulations with millions of molecules are computationally demanding. To simulate a typical system on the scale of several hundred nanometers for a time length of only a few nanoseconds, it would require a today’s desktop computer to run for many years. Thus, supercomputers are utilized in order to massively accelerate such simulations. The open source code ls1 mardyn [1,2], which was used to compute the scenarios that are described in the following, is designed for the use on supercomputers and thus can be run in massively parallel execution mode. In the following scenarios, MD simulations yield insights in the context of fluid mechanics and help to improve modelling with CFD simulations. The illustrations were rendered by using the open source visualization framework MegaMol [5]
Shock wave propagation
Shock wave propagation plays an important role when it comes to the development of new methods in the context of fluid mechanics since it is one manifestation of the Riemann problem. The Riemann problem is an initial value problem in which all initial values are constant besides one discontinuity. It is challenging to realize such a case with the required accuracy by experiments. Also, the conduction of DNS is difficult because of the system’s size of a few hundred nanometers so that MD was utilized. Continuing preceding work [3], in the present scenario [4] a slab of a liquid phase is placed next to a vapor phase. This leads to the aforementioned Riemann problem and non-equilibrium effects occur at the phase boundary. For technical reasons and to improve the sampling accuracy, the system was mirrored, hence resulting in a liquid phase surrounded by two vapor phases as is can be seen in Fig. 1.
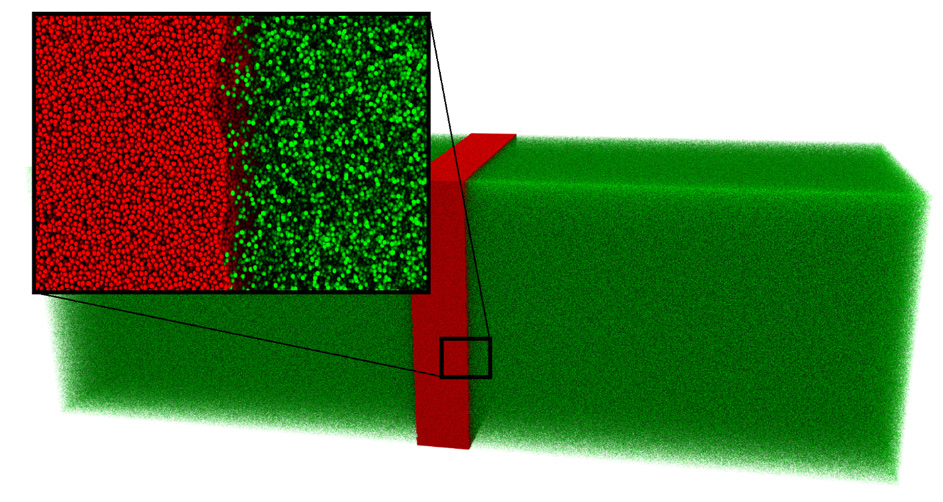
Figure 1: Simulation system consisting of two vapor phases (green) and one liquid phase (red). Single atoms can be identified in the marked scope which magnifies the phase boundary. © TU Berlin
The overall simulation system contained about 300 million particles, which is why about 10,000 cores were needed to run simultaneously for approximately 20 hours in order to perform the simulation. When the simulation is started, evaporating liquid induces a shock wave propagating into the vapor. During the run, the temporal evolution of temperature and density profiles were sampled. The results of the simulation were compared to data obtained by two different macroscopic numerical methods as they are used in CFD simulations. It turned out that one of the numerical methods was not able to predict the correct fluid behavior, while the other method showed a great agreement with the MD data.
Droplet coalescence
Another scenario in which MD simulations results are used to validate macroscopic numerical methods is droplet coalescence. Since MD is capable of not only predicting thermodynamic properties but also investigating the hydrodynamic behavior of fluids, it excels in this scenario. Two droplets with a diameter of 50 nanometers, respectively, were positioned next to each other. The whole system consisted of about 3 million atoms. As soon as the simulation starts, the two droplets attract each other and coalesce. The overall process is highly dynamical and takes about 10 nanoseconds. A snapshot of the system can be seen in Fig. 2.
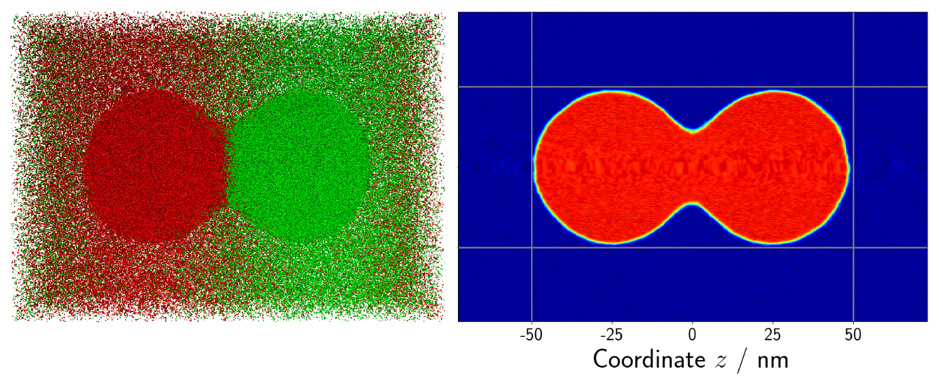
Figure 2: Snapshot of two coalescing droplets. Left: The colors indicate the affiliation of the respective atom to the initial droplet. Right: Density map of the left configuration. Regions with high density (liquid) are colored in red, blue indicates low density (vapor). © TU Berlin
Injection
Injections are an omnipresent phenomenon. The first field to think of are engines, but injections are also important in many other fields. Analogous to the drop coalescence, MD simulations are capable of predicting the structure of the fluid during injection processes. Figure 3 (as well as the mp4 video below) shows an exemplary injection of a monatomic liquid, where vaporization as well as formation of droplets was observed. This formation was not modelled in advance, but is solely a result of the interactions between the atoms. Therefore, this scenario impressively demonstrates again that MD simulations excel in situations where the prediction of the thermo- and hydrodynamic behavior of fluids and the validation of methods used in CFD simulations is of major interest.

Figure 3: Snapshot of the injection of a monatomic liquid into vacuum. © TU Berlin
mp4 video: Video of the injection of a monatomic liquid into vacuum. © TU Berlin
References
[1] C. Niethammer, S. Becker, M. Bernreuther, M. Buchholz, W. Eckhardt, A. Heinecke, S. Werth, H.-J. Bungartz, C. W. Glass, H. Hasse, J. Vrabec, M. Horsch. ls1 mardyn: The Massively Parallel Molecular Dynamics Code for Large Systems, Journal of Chemical Theory and Computation10 (2014) 4455-4464. doi:10.1021/ct500169q
[2] N. Tchipev, S. Seckler, M. Heinen, J. Vrabec, F. Gratl, M. Horsch, M. Bernreuther, C. W. Glass, C. Niethammer, N. Hammer, B. Krischok, M. Resch, D. Kranzlmüller, H. Hasse, H.-J. Bungartz, P. Neumann. Twetris: Twenty trillion-atom simulation, The International Journal of High Performance Computing Applications 33 (2019) 838–854. doi:10.1177/1094342018819741
[3] T. Hitz, M. Heinen, J. Vrabec, C.-D. Munz. Comparison of macro- and microscopic solutions of the Riemann problem I. Supercritical shock tube and expansion into vacuum, Journal of Computational Physics 402 (2020) 109077. doi:10.1016/j.jcp.2019.109077
[4] T. Hitz, S. Jöns, M. Heinen, J. Vrabec, C.-D. Munz. Comparison of macro- and microscopic solutions of the Riemann problem II. Two-Phase Shock Tube, Journal of Computational Physics, in press (2021). doi: 10.1016/j.jcp.2020.110027.
[5] S. Grottel, M. Krone, C. Müller, G. Reina, and T. Ertl, IEEE Trans. Visualization Comput. Graphics 21 (2015) 201-214. doi:10.1109/TVCG.2014.2350479
Research Team
Matthias Heinen, Simon Homes, Jadran Vrabec (PI).
All: Technische Universität Berlin, Chair of Thermodynamics and Process Engineering
Scientific Contact
Prof. Dr.-Ing. Jadran Vrabec
Chair of Thermodynamics and Process Engineering
Technische Universität Berlin
Ernst-Reuter-Platz 1, D-10587 Berlin (Germany)
e-mail: vrabec [@] tu-berlin.de
Local project ID: MMHBF2
February 2021