
MATERIALS SCIENCE AND CHEMISTRY
Modelling the Mechanisms of Complex Nanocatalysts
Principal Investigator:
Maddalena D'Amore
Affiliation:
Department of Chemistry, University of Turin
Local Project ID:
pr27si
HPC Platform used:
SuperMUC of LRZ
Date published:
Heterogeneous catalysts have a complex nature that makes it difficult to determine atomic-level knowledge of their active sites, especially in reaction conditions. Traditional experimental methods are only able to provide a partial picture of how these catalysts function.
Now, due to a combination of developments in DFT (Density Functional Theory) and the availability of powerful computational facilities, it is starting to become possible to design efficient heterogeneous catalysts from first principles. The NANOCAT-ZN project, led by Dr. Maddalena D’Amore at the Department of Chemistry of the University of Turin, is using an allocation on the SuperMUC machine hosted by GCS at LRZ, Germany, to model the mechanism of reactions and active sites in Ziegler-Natta nanocatalysts.
Ziegler-Natta catalysts are an important class of mixtures of chemical compounds that are well known for their ability to polymerise “olefins” (hydrocarbons containing a double carbon–carbon bond) into polymers of high molecular weights and highly ordered structures; they were developed in the 1950s by German chemist Karl Ziegler whereas Giulio Natta extended the research conducted by Ziegler on organometallic catalysts to the stereospecific polymerization, thus discovering new classes of polymers with a sterically ordered structure. These complex nanometre-sized materials, composed of MgCl2, TiCl4, an aluminium alkyl and organic Lewis bases, are computationally demanding systems to investigate, as D’Amore explains. “To understand how Ziegler-Natta catalysts work, you must take into account a number of things. Firstly, there are multiple active sites on different MgCl2 surfaces and on defective positions originated by both disorder and mostly nanostructuration phenomena. Secondly, the chemical compounds of catalytic interest – involving TiCl4, aluminium alkyls and very bulky aromatic donors – have a very low degree of coverage. This is why it is necessary to model very large systems that are very far from perfection to achieve a reliable description of catalysts.“
“Finally, after the reduction of titanium species, unrestricted DFT calculations are necessary. For these reasons, an accurate quantum-mechanical prediction of the thermodynamics and kinetics of these systems requires highly expensive calculations and huge computational resources.“
The team led by D’Amore used an efficient highly parallelised code called MPPCRYSTAL to carry out DFT simulations, correlating the structure of the Ziegler-Natta catalysts at the nanoscale with their activity and selectivity to understand the role of each component in the reaction mixture. The code was specifically tailored to achieve an excellent scalability in terms of both speed-up and memory usage, allowing the researchers to treat very large unit cell systems with large memory requirements and few or no symmetry operators. The project has been truly multidisciplinary, representing a collaboration between theoreticians, spectroscopists and industrial partners. Their joint aim was to find out more about how the structure of the active sites in these catalysts contribute to their function and the role of “each player” and synthetic procedure in the game. “As well as this, we wanted to both improve the industrial performance of these catalysts and the performance of our code when running on thousands of processors,” says D’Amore.
The simulations of the catalysts used models that involved hundreds of atoms – a huge amount in the world of accurate DFT. Due to the large size of the investigated systems and the high level of accuracy of calculations, that employs both hybrid exchange-correlation functionals and extended basis sets, the feasibility of simulations was possible only thanks to the HPC resources granted by PRACE on HPC system SuperMUC.
The realistic description of these catalysts required advanced DFT methods accounting for dispersion and employing hybrid functionals or highly parameterised m-GGA exchange-correlation functionals to describe metal catalytic sites. Thanks to the use of thousands of cores simultaneously, it was possible to simulate nano-crystals of up to one thousand atoms, analysing plausible reactivity indicators such as electron density transfers in order to evaluate the possible occurrence of specific reaction mechanisms, on the basis of ELF and MPD (Maximum Probability Domains) theories.
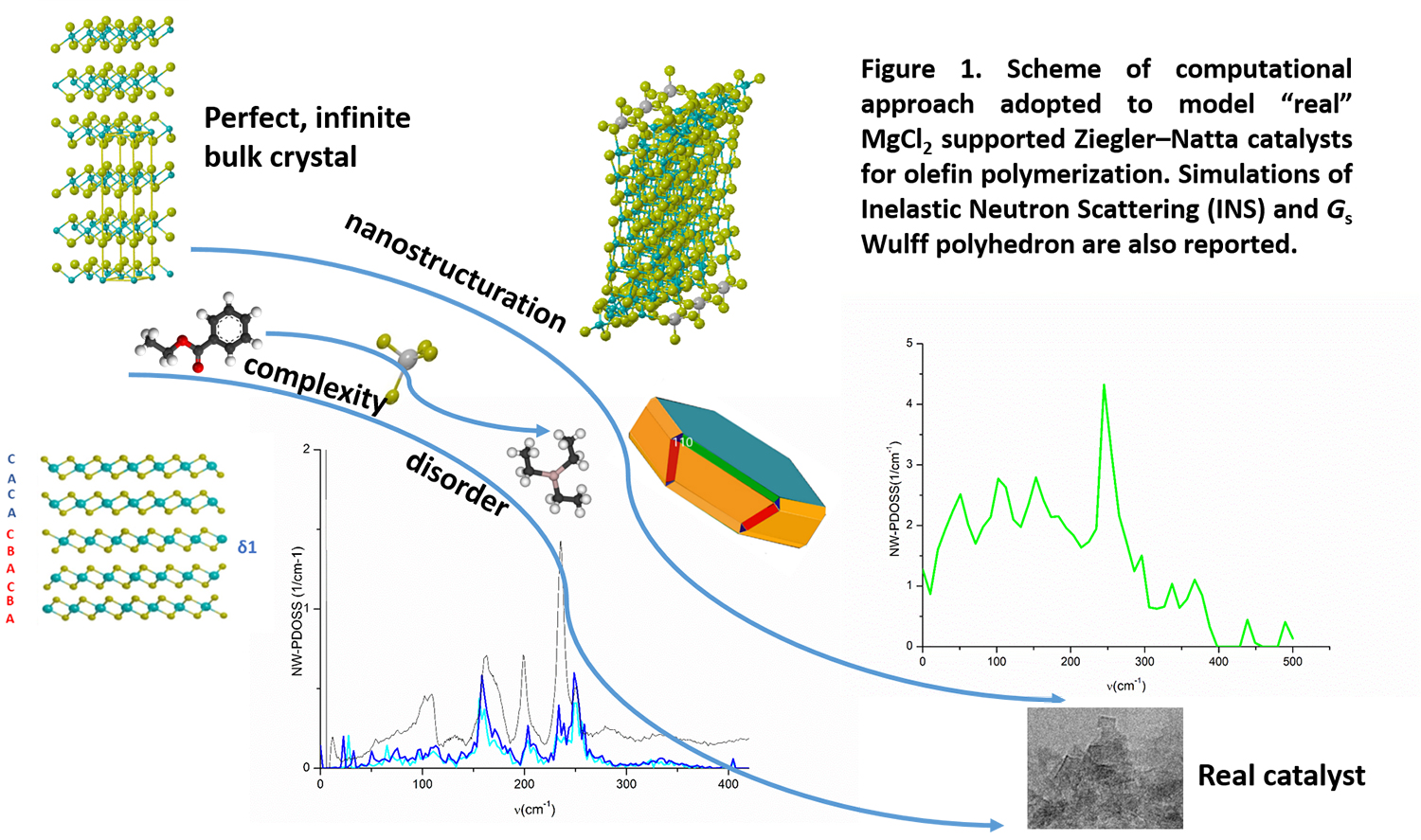
The investigation modelled low-dimension systems: surfaces, nano-rods and energetically stable primary particles, adopting (when periodicity is still kept) a supercell approach to build huge unit cells to simulate adducts with poor loadings (Figure 1). Thanks to vibrational analysis, the entropic contribution to surface formation energy was estimated in reaction conditions. “To our surprise, the (110) surfaces, identified by recent theoretical models as the place for stereo-selective active sites of catalytic processes involving TixCly species, arose on our simulated Wulff polyhedron,” says D’Amore. “This is the first time that the active catalytic surface has emerged in a quantum mechanical simulation in reaction conditions.”
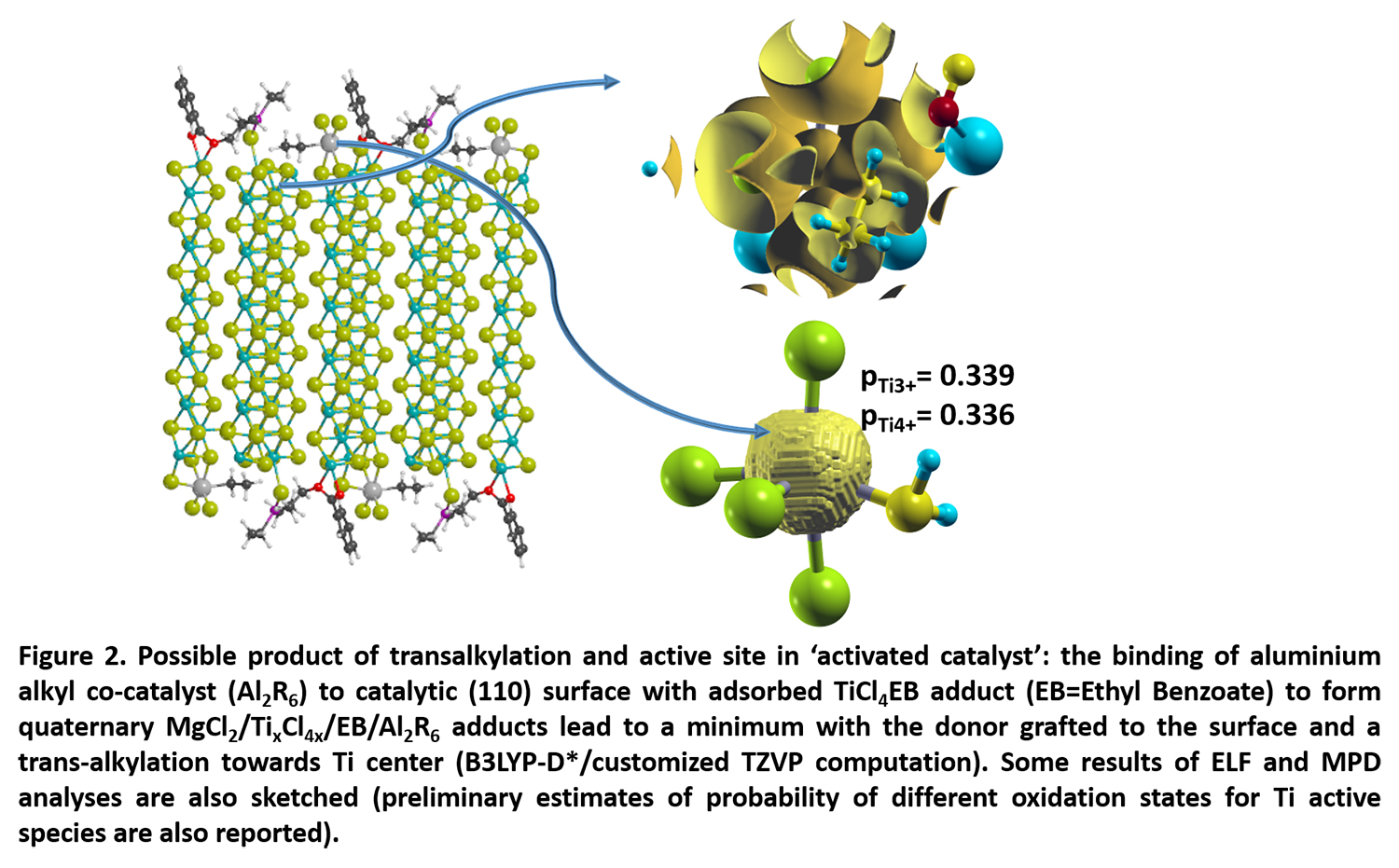
These results of the project have helped to contribute to the understanding of the relation between the structure, reactivity and selectivity of the catalysts’ active sites towards olefins. Starting from the identified precatalytic sites and assuming that the adjacent donors affect the electron density of metal active sites, D’Amore and her team plan to continue investigating the mechanism of titanium reduction and olefin insertion starting from identified quaternary adducts (i.e. Figure 2). In this way, it could well be possible to work out how to improve the productivity and stereospecificity of these catalysts once a better understanding of the process by which they function is achieved, making the “trial and error” method used by industry obsolete.
At the same time, this will help to reduce the health and environmental impacts that Ziegler-Natta catalysts can have by increasing the efficiency of these catalysts and also hypothesize and identify future more 'green and healthy' compounds in the working mixture. The team used poorly validated theoretical methodologies as MPD to surface catalysis; (Figure 2) the application of these methodologies in other fields of heterogeneous catalysis would be a valuable and long-term contribution of the project out of the specific scientific field of polymer catalysis.
Publications
“A Periodic Hybrid DFT Approach (Including Dispersion) to MgCl2-Supported Ziegler-Natta Catalysts, 1. TiCl4 Adsorption on MgCl2 Crystal Surfaces”, Journal of Catalysis, 2012.
”Surface Investigation And Nanoscale Morphological Analysis Of Coordinatively Unsaturated Surfaces In Structurally Disordered MgCl2 And MgCl2/TiCl4 Ziegler-Natta Catalysts”, ACS Catalysis, 2016, 6, 5786-5796.
“The Bond Analysis Techniques (ELF and Maximum Probability Domains) Application to a Family of Models Relevant to Bio-Inorganic Chemistry”, in “Applications of Density Functional Theory to Biological and Bioinorganic Chemistry”. Series: Structure and Bonding, Vol. 150.
“Periodic and High-Temperature Disordered Conformation of Polytetrafluoroethylene Chains: an Ab-Initio Modelling”, Journal of American Chemical Society 2006,128(4); 1099-1108.
T. Wada, G. Takasao, A. Piovano, M. D'Amore, A. Thakur, Patchanee Chammingkwan, P. C. Bruzzese, M. Terano, B. Civalleri, S. Bordiga, E. Groppo, and T. Taniike "Revisiting the identity of δ-MgCl2: Part I. Structural disorder studied by synchrotron X-ray total scattering". Journal of Catalysis, accepted.
A. Piovano, M. D'Amore,* T.Wada, P. C. Bruzzese, G. Takasao, A. Thakur, P. Chammingkwan, M. Terano, B. Civalleri, S. Bordiga, T. Taniike, E. Groppo. "Revisiting the identity of δ-MgCl2: Morphology and exposed surfaces studied by vibrational spectroscopies and DFT calculation". Journal of Catalysis, submitted.
Scientific Contact
Dr. Maddalena D’Amore
Department of Chemistry
Università degli Studi di Torino
Via P. Giuria, 7 35 10125, I-Torino (Italy)
e-mail: maddalena.damore [@] unito.it
http://www.crystal.unito.it/index.php
NOTE: This is a reprint of the article published in PRACE Digest 2019, p.18-19. The simulation project was made possible by PRACE (Partnership for Advanced Computing in Europe) allocating a computing time grant on GCS HPC system SuperMUC of the Leibniz Supercomputing Centre (LRZ) in Garching near Munich. GCS is a hosting member of PRACE.
LRZ project ID: pr27si
March 2020