
MATERIALS SCIENCE AND CHEMISTRY
Theoretical Infrared Spectroscopy of Solvated Biomolecules from ab initio Molecular Dynamics: Understanding Aqueous TMAO Solutions at Very High Pressures
Principal Investigator:
Dominik Marx
Affiliation:
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum (Germany)
Local Project ID:
pr23va
HPC Platform used:
SuperMUC of LRZ
Date published:
Organisms which live under extreme conditions have established adaptation mechanisms during their evolution. One such mechanism with enables life under kbar pressures in deep sea habitats is an unusually high concentration of a specific molecule in their blood, namely TMAO (trimethylamine N-oxide). Yet, the so-called piezolytic mechanism which counteracts such high pressure effects within cells is not understood. As a first step, scientists investigated the properties of aqueous TMAO solutions at very high pressure compared to ambient conditions and find significant changes in the hydrogen bonding properties.
The Challenge
A lot of organisms on earth live under extreme conditions. For example, deep sea creatures survive under pressures above 100 bar. A myriad of microorganisms even lives under much higher pressure conditions, e.g. below sub-seafloor, and enter the kbar regime. TMAO (trimethylamine N-oxide) is known to counteract the pressure-induced denaturation of proteins and, incidentally, is found in deep sea fish in concentrations which increase strongly with decreasing average habitat depth of the animal as shown by experimental biologists. Despite significant research into TMAO/protein interactions at ambient conditions, close to nothing is known about such solutions at compression into the kbar range. This study is a first step towards deciphering the solvation properties of TMAO in water from ambient conditions up to 10 kbar at 300 K.
Necessity for HPC Facility
Molecular dynamics simulations are a well-established tool to analyze molecular details of complex aqueous systems. It is, however, not at all obvious whether commonly used force fields are applicable when moving much beyond ambient conditions where they have been carefully parameterized and validated. One way to tackle the issue is to develop pressure-dependent force fields [1], which however is a rather long-term strategy. Immediate access is provided by ab initio simulations [2] in which the inter-atomic interactions are computed from concurrent electronic structure calculations which is readily and accurately applicable from ambient to extreme conditions without any adjustments.
On the other hand, even state-of-the-art ab initio simulation software packages such as CP2k [3] require enormous compute resources to enable realistic applications. In particular infrared spectra, which are based on time-correlation functions, require a massive amount of statistics in order to quantitatively converge the intensities and thus the lineshape function, whereas the simulated system by itself is rather "small". The SuperMUC system turned out to be the ideal platform to carry out these simulations.
Results
Characteristic to the TMAO molecule (see insets in the figure) are strongly hydrophilic and bulky hydrophobic groups at the opposite ends as well as its large dipole moment. Especially the strong hydrophilic oxygen site accepts about three hydrogen bonds from nearby water molecules, whereas this number increases systematically upon compression to 10 kbar [4].
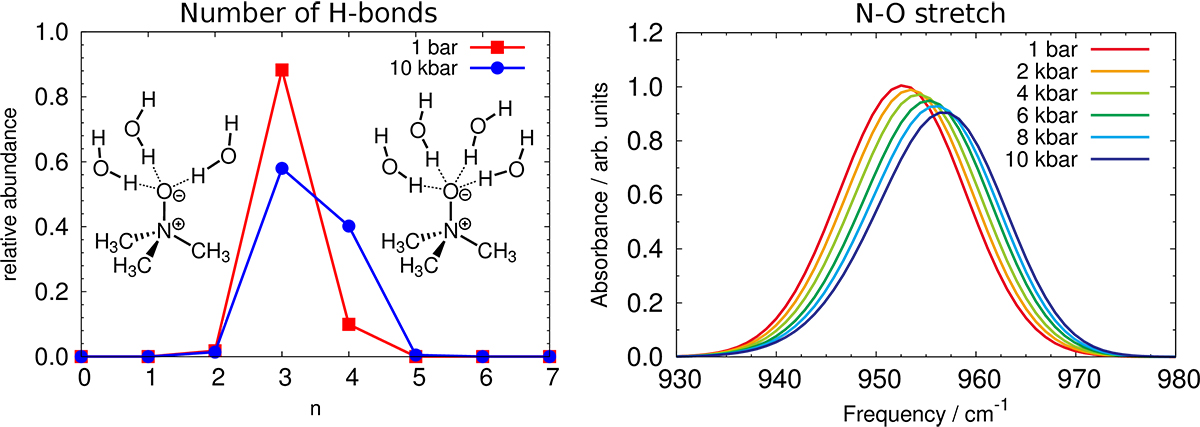
(Left Panel) Number of hydrogen bonds between TMAO and water at 1 bar (red) and 10 kbar (blue); the insets show schematically the structures of the two relevant supermolecular solvation complexes, namely TMAO(H2O)3 (left) and TMAO(H2O)4 (right). (Right Panel) Pressure dependence of the N-O stretching peak (see [5] for background and method).
Copyright: Ruhr-Universität Bochum (Germany)Analysis has shown that a solvated TMAO can be considered as a TMAO(H2O)3 complex at 1 bar, whereas a mixture of TMAO(H2O)3 and TMAO(H2O)4 is found in the solution at 10 kbar (see left panel in the figure). At the first glace, this pressure response seems very natural because high pressure conditions in general result in shorter intermolecular distances. The number of hydrogen bonds between water molecules in pure water at 300 K, however, has shown to be almost constant from 1 bar to 10 kbar [4].
Thus, the puzzle is to understand, at the molecular level, the stark difference of the pressure response between aqueous TMAO solutions and pure liquid water. The researchers found a strong correlation between the magnitude of the TMAO dipole moment in solution and the number of hydrogen bonds between TMAO and water, since hydrogen bonding water molecules pull electrons away from the TMAO oxygen. The increases in the TMAO dipole moment and the number of hydrogen bonds occur in a correlated manner and, indeed, additional force field simulations revealed that an enhanced TMAO dipole moment is the key to describe the characteristic change of the hydrogen bonding pattern of TMAO in water upon compression [1]. In contrast, the dipole moment of the water molecules remains almost constant from ambient up to 10 kbar and, therefore, the number of hydrogen bonds between water does not change upon compression.
Last but not the least, there was the question as to how to connect the findings to experimental measurements? To elucidate the change in the hydrogen bond pattern around TMAO oxygen, the scientists focused on the N-O stretch mode in the infrared spectrum (see right panel of the figure and [5]).
The resonance of the TMAO(H2O)4 complexes appears at slightly lower frequencies with respect to TMAO(H2O)3 since more hydrogen bonds result in a weaker N-O bond. The ratio of TMAO(H2O)4 increases upon compression and, thus, the N-O stretch shows an asymmetric tail to the low frequency side at HHP conditions. Indeed, both experimental and simulated infrared spectra show the increase in skewness at high pressure conditions [5]. This agreement clearly supports the prediction [3] based on ab initio simulations on how the solvation properties of aqueous TMAO solutions change when moving into the realm of high pressures.
Outlook
Future work will be devoted to urea since that particular molecule is known to destabilize folded proteins. Again, its solvation properties at high pressures are terra incognita but can be explored readily using ab initio molecular dynamics.
Acknowledgements
This project has been partially funded by the Deutsche Forschungsgemeinsschaft in terms of the Research Unit "Exploring the Dynamical Landscape of Biomolecular Systems by Pressure Perturbation" (FOR 1979) and is also part of the Cluster of Excellence “RESOLV” (EXC 1069).
References
[1] Christoph Hölzl, Patrick Kibies, Sho Imoto, Roland Frach, Saba Suladze, Roland Winter, Dominik Marx, Dominik Horinek and Stefan M. Kast, “Design principles for high–pressure force fields: Aqueous TMAO solutions from ambient to kilobar pressures”, J. Phys. Chem. 114, 144104 (2016).
[2] Dominik Marx and Jürg Hutter, “Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods” Cambridge University Press, Cambridge, (2009).
[4] Sho Imoto, Harald Forbert and Dominik Marx, “Water structure and solvation of osmolytes at high hydrostatic pressure: pure water and TMAO solutions at 10 kbar versus 1 bar”, Phys. Chem. Chem. Phys. 17, 24224 (2015).
[5] Sho Imoto, Patrick Kibies, Christopher Rosin, Roland Winter, Stefan M. Kast and Dominik Marx, “Toward Extreme Biophysics: Deciphering the Infrared Response of Biomolecular Solutions at High Pressures”, Angew. Chem. Int. Ed. 55, 9534 (2016).
Scientific Team:
Prof. Dr. Dominik Marx (PI) and Dr. Sho Imoto
Scientific Contact:
Prof. Dr. Dominik Marx
Lehrstuhl für Theoretische Chemie - Ruhr-Universität Bochum
Universitätsstraße 150, D-44801 Bochum (Germany)
e-mail: dominik.marx[at]rub.de
October 2017