
MATERIALS SCIENCE AND CHEMISTRY
Transition Metal Carbides for Electrochemical CO2 Reduction
Principal Investigator:
Karsten Reuter
Affiliation:
Lehrstuhl für Theoretische Chemie, Technische Universität München
Local Project ID:
tmcscat
HPC Platform used:
JUWELS of JSC
Date published:
Introduction
Electrochemical CO2 reduction is a clean and promising approach to reduce the amount of CO2 in the atmosphere and fight climate change. Required is the development of highly selective and efficient catalysts for CO2RR that generate targeted valuable chemical and fuel products. Limited by linear scaling relationships between the adsorption strengths of reaction intermediates to the catalyst surface, the protonation of adsorbed *CO intermediates to *CHO is the potential determining step across transition metal (TM) cataysts. Even the optimal metal catalyst Cu still tends to generate hydrocarbons with a high overpotential of ~ 1V. Future catalysts should thus strengthen the binding energy of *CHO relative to *CO at the surface.
Transition metal catalysts (TMCs) are promising to break the scaling relationship through the wide variety of structural and compositional motifs exhibited on their surfaces (Figure 1) and thereby achieve higher catalytic performance. Analyzing the adsorption strength of a range of key CO2RR reaction intermediates toward methane and methanol, many of the scaling relations known from transition-metal adsorption were found to be strongly broken across the different sites. This highlights a rich reduction chemistry at the carbides beyond what has been optimized for Cu. In particular, a selectively weakened *CO adsorption relative to *CHO helps to overcome the limitations for CO2RR imposed by the strong correlation of the two adsorption energies at transition metals. In addition to the hitherto heralded suitability for CO2RR to methane, our calculations suggest carbon-rich active sites at both MoC and Mo2C to also generate methanol at low overpotentials.
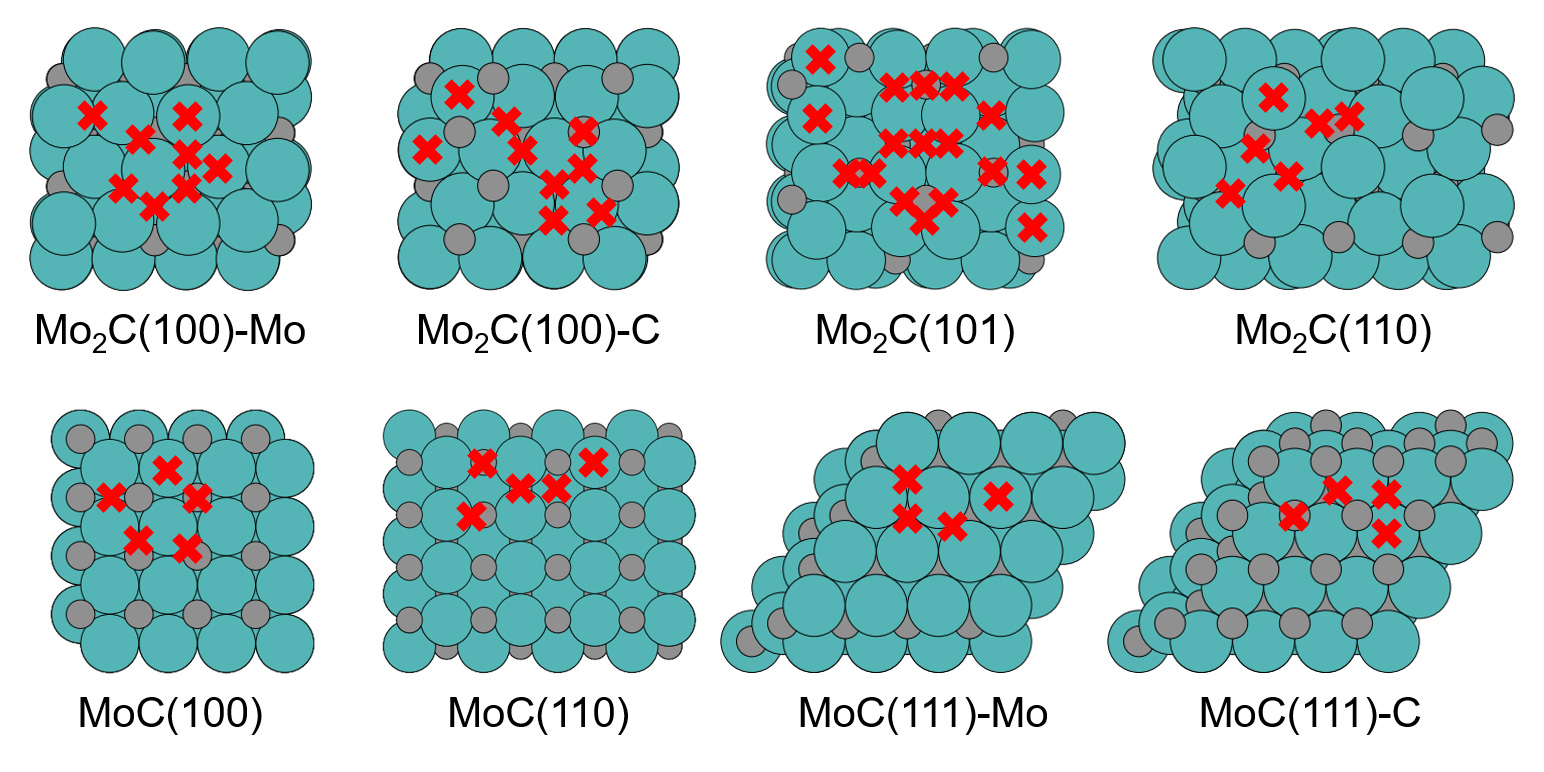
Figure 1. Top views of the considered set of eight low-index facets and surface terminations of orthorhombic Mo2C and cubic MoC (Mo = large green spheres, C = small gray spheres). The resulting 58 nominal high-symmetry sites are marked by red crosses. © TUM
Results
The study of Mo carbides is based on two crystal phases with different Mo/C ratios, namely, orthorhombic Mo2C and cubic MoC. For the considered surface adsorption species *CO2, *COOH, *CO, *CHO, *CH2O, *CHOH, *CH3O, *CH2OH, *O and *OH, a full structural optimization has been carried out on each high-symmetry site of the eight surfaces. The unstable sites were discarded, and a dataset of adsorption free energies has been generated. An analysis of the data shows that the C adsorption species are divided into two different adsorption modes, one retains the up-right adsorption mode for *CO and *CO2, and the other prefers a side-on configuration on the surface for *COOH, *CHO, *CH2O, *CHOH and *CH2OH. The linear scaling relationship is broken between different adsorption modes, while it still exists between adsorbates in the same adsorption mode. The consequences are prominently illustrated for the adsorbates *CO and *CHO involved in the potential-determining CO2RR reaction step on TMs. The diversity of the active-site structures leads to different adsorption modes, which affects the relationship between the adsorption free energies, and thus affects the activity and selectivity of the reaction.
Within the minimalistic thermodynamic reaction network used in Figure 2, it is intriguing to see that for the majority of sites, the actual *CO protonation sequence to a C1 product is not really the true bottleneck. Definitely for Mo-rich sites which generally exhibit rather strong *OH binding, the UL value for this desorption step is much more negative than the most reducing UL for the protonation sequence, and correspondingly, the regeneration of these adsorption sites for the next reaction cycle is the truly potential-limiting step. Another striking trend is that all C-rich sites lead unanimously to methanol, whereas all Mo-rich sites lead to methane. From a different angle, this confirms the importance of the metal/carbon ratio and the suitability of C-rich active-site motives to generate CH3OH.
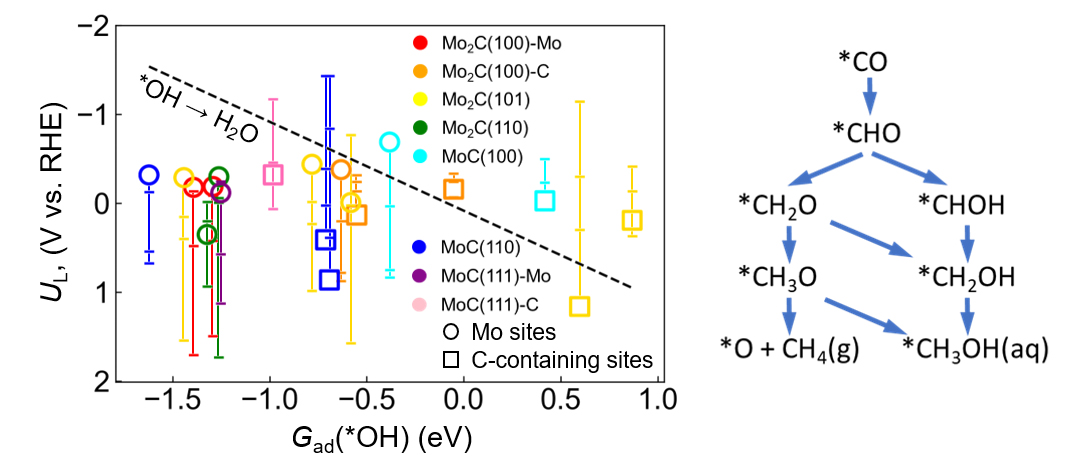
Figure 2. Limiting potentials UL (on the RHE scale) below which individual *CO protonation steps become exergonic across different MoxC (x = 1, 2) active sites. For each active site, the vertical line spans the UL for the four PCET (proton-coupled electron transfer) steps following the most exergonic path to either CH4 or CH3OH within the minimalistic reaction network shown on the right. © TUM
Acknowledgments
The project team members received funding through the Alexander von Humboldt (AvH) foundation and the German Research Council (DFG) through project RE1509/36-1.
References and links
[1] Li, H.; Reuter, K. ACS Catal. 10, 11814–11821 (2020). https://pubs.acs.org/doi/abs/10.1021/acscatal.0c03249
[2] Griesser, C.; Li, H.; Wernig, E.-M.; Winkler, D.; Nia, N. S.; Gotsch, T.; Schachinger, T.; Steiger-Thirsfeld, A.; Penner, S.; Wielend, D.; Egger, D.; Scheurer, C.; Reuter, K.; Kunze-Liebhauser, J. (in revision).
Research team
Haobo Li, Karsten Reuter (PI)
Lehrstuhl für Theoretische Chemie, Technische Universität München, Germany
https://www.department.ch.tum.de/th4/lehrstuhl-fuer-theoretische-chemie/
Scientific Contact
Dr. Haobo Li
Lehrstuhl für Theoretische Chemie
Technische Universität München, Germany
Lichtenbergstraße 4, D-85747 Garching/Munich (Germany)
e-mail: haobo.li [@] tum.de
Local project ID: tmcscat
March 2021