
MATERIALS SCIENCE AND CHEMISTRY
Ab Initio Modeling of Materials for and by Georesources Under High Pressure
Principal Investigator:
Prof. Dr. Maribel Núñez-Valdez
Affiliation:
Helmholtz-Zentrum Potsdam
Local Project ID:
abinitiomodmatsgeo
HPC Platform used:
JUWELS at JSC
Date published:
Introduction
The behavior and properties of materials under extreme compression/pressure often change drastically. For example, changes of symmetry and atomic arrangement (phase transformations) can occur, superconductivity might appear, magnetic ordering can change, etc. Experimentally, researchers have developed a wide range of techniques, often based on the diamond anvil cell (DAC) to study the pressure effects on a variety of materials of interest in solid state physics, chemistry, materials sciences, geophysics, planetary sciences and astronomy. However, due to the challenges presented by in-situ observations, frequently the only other possible method available is first-principles modeling.
The first-principles or ab initio approach employed in these investigations is based on quantum mechanical electronic codes that predict, and facilitate the understanding and interpretation of a material’s properties under extreme pressure and/or temperature. The general method uses density functional theory (DFT) [1,2] and does not require experimental input aside of the constituent atoms, and perhaps, a starting crystal structure of the material under study. First-principles outcomes are important in the landscape of the development of new applications, and the role of modeling is imperative as it helps in designing experiments to test a prediction or provides predictions when laboratory measurements are nearly impossible to achieve.
The structural, electronic and magnetic properties of some (geo)materials, as well as their intricate correlations, lie currently at the center of scientific attention worldwide, owing to the large variety of parameters affecting them, such as composition, symmetry, stoichiometry, as well as temperature and pressure conditions. Notably, over the last few decades and in recent years, classic cubic semiconductors are continuously inspiring and fueling the search for novel properties.
Results
Tailoring materials with specific properties to extend their functionality is often impossible under standard conditions of pressure and temperature. For instance, Si and Ge, with a diamond structure (cubic symmetry) at ambient pressure, are the most used materials in the semiconducting industry and the desire for their applications not only in microelectronics but also in optoelectronics has generated new research such as doping, etching, and tensile strain of diamond-(Si,Ge), in order to overcome their indirect band gap hurdle. Therefore, a novel approach inspired by experimental high-pressure studies was to use pressure to transform the diamond-(Si,Ge) and develop a new SixGe1-x landscape that contains tunable properties such as the nature of the band gap [3]. Experimentally, solid solutions with cubic and tetragonal crystal symmetries were synthesized with promising electronic character as semiconducting, semimetallic, thermoelectric, optoelectronic materials. Thus, with our computing simulations using JUWELS, we investigated SixGe1-x alloys in the entire compositional range (0<x<1) in their metastable tetragonal (ST12) and cubic (BC8) symmetries.
To analyze order/disorder effects on phase stability and lattice parameters, we modeled all possible atomic site occupancies in the primitive cells of ST12 and BC8. By taking into account space group symmetry, we investigated a total of 362 configurations in the ST12 modification (12 atoms /cell), and a total of 475 in the BC8 modification (8 and 16 atoms/cell). As a second step, we modeled randomly distributed supercells for intermediated compositions, as the experimentally synthesized alloys are expected to be fully disordered. We achieve this by generating special quasi random structures (SQS) of 96-atom supercells.
With our general approach, we were able to calculate the stability field, lattice parameters and band properties for intermediate compositions of both BC8 and ST12 solid solutions. Our results indicated two important predictions for the alloy lattice parameters (Fig. 1), the first is a weighted average according to the symmetry derived probability of all primitive cell configurations for a particular composition x, and the second is directly derived from the SQS supercell optimizations. Overall, our results showed excellent agreement with experimental findings and approximately follow the Vegard’s law.
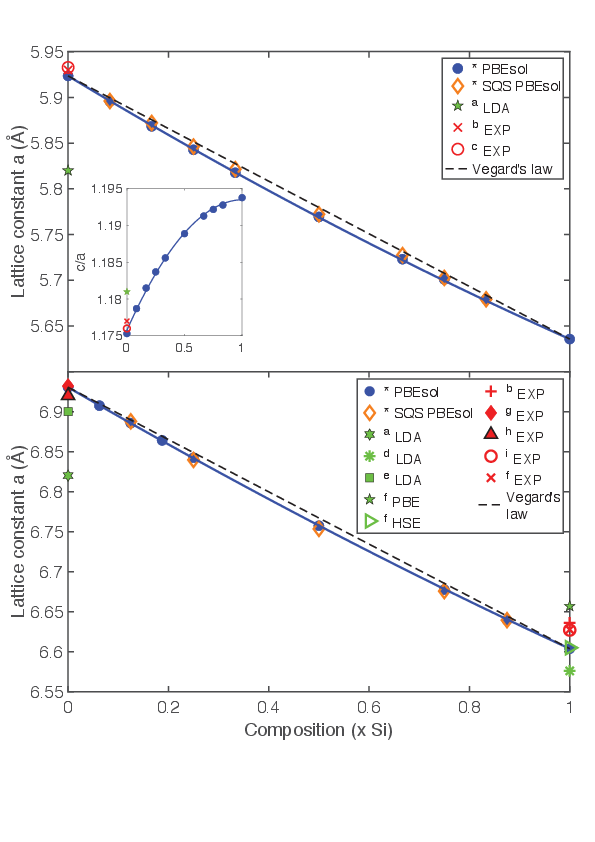
Figure 1. Lattice constant a and bowing curve for ST12 (top) and BC8 (bottom) SixGe1-x alloys. Blue dots are the averaged value of all possible site occupancy configurations for one individual composition x. The inset shows the c/a ratio for all ST12 alloys. Orange diamonds are the lattice parameters directly derived from special quasi-random structures (SQSs). Theoretical error bars are smaller than symbols. Our results are marked by *. References for data of other simulations and experiments can be found in our publication [WNV].
In relation to the electronic band structure in the ST12 modification, a slight addition of Si to pure ST12 Ge resulted in a shift of the valence band maximum (VBM) towards the position of the conduction band minimum (CBM), culminating in a direct band gap at approximately xSi≈0.16 (Fig. 2B). Adding more Si resulted in the VBM shifting entirely to produce once again an indirect band gap.
On the other hand, we found that BC8 Si is a narrow gap semiconductor only at end member composition. Adding a little of Ge (xGe= 0.06 in our study) resulted in a metallic nature of the alloy. Further details and all results derived from this study can be found published in the Applied Physics Letters journal [WNV].
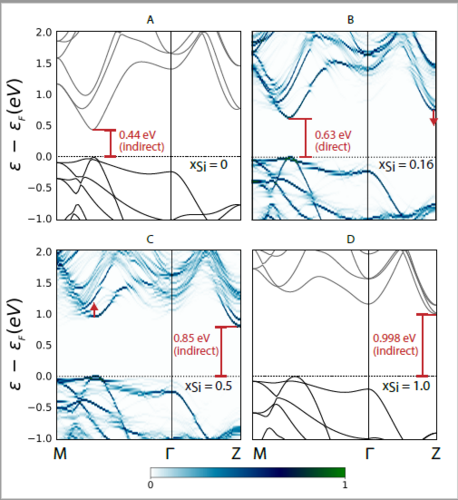
Figure 2. ST12 alloy band structure plots (A) and (D) are plots derived from the primitive end-member simulation cells, (B) and (C) show effective unfolded band structures derived from special quasi-random structures (SQSs). (A) Pure ST12 Ge with a well-studied indirect bandgap. (B) Effective SQS band structure for xSi≈0.16 showing a direct fundamental (C) Effective SQS band structure for x=0.5 where the original CBM has shifted right, resulting in a large indirect bandgap. (D) Band structure of pure hypothetical ST12 Si.
Publication
[WNV] J. Wagner and M. Núñez Valdez, Ab initio study of band gap properties in metastable BC8/ST12 SixGe1-x alloys, Appl. Phys. Lett. 117, 032105 (2020)
https://doi.org/10.1063/5.0010311
References
[1] P. Hohenberg and W. Kohn, Phys. Rev., 136, B864–871 (1964)
https://doi.org/10.1103/PhysRev.136.B864
[2] W. Kohn and L. J. Sham, Phys. Rev., 140, A1133–1138 (1964)
https://doi.org/10.1103/PhysRev.140.A1133
[3] G, Serghiou, Ji, G., Koch-Müller, M., Odling, N., Reichmann, et al., Inorg. Chem., 53, 11, 5656-5662 (2014)