
MATERIALS SCIENCE AND CHEMISTRY
Ab Initio Modelling of the Adsorption in Giant Metal-Organic Frameworks: From Small Molecules to Drugs
Principal Investigator:
Bartolomeo Civalleri
Affiliation:
Dept. of Chemistry, University of Torino (Italy)
Local Project ID:
pr85qu
HPC Platform used:
SuperMUC of LRZ
Date published:
Metal-Organic Frameworks (MOFs) are a new class of materials that in the last decade have seen a paramount growth and are expected to have huge impact on the development of next-generation technologies. They consist of inorganic nodes (i.e. a metal ion or a cluster) connected through organic linkers to form a porous 3D framework. The combination of different nodes and linkers makes MOFs very versatile materials with interesting and promising applications in many fields, including: gas adsorption, catalysis, drug delivery, nonlinear optics.
This project has focused on the ab initio modeling of the adsorptive capacity of the so-called giant MOFs, in particular of MIL-100 (see Figure 1) because of the very large size of the pores (meso-pores: 25–30 Å) and a corresponding huge surface area (3,340 m2 g−1) which is three times larger than for commonly used adsorbent materials.
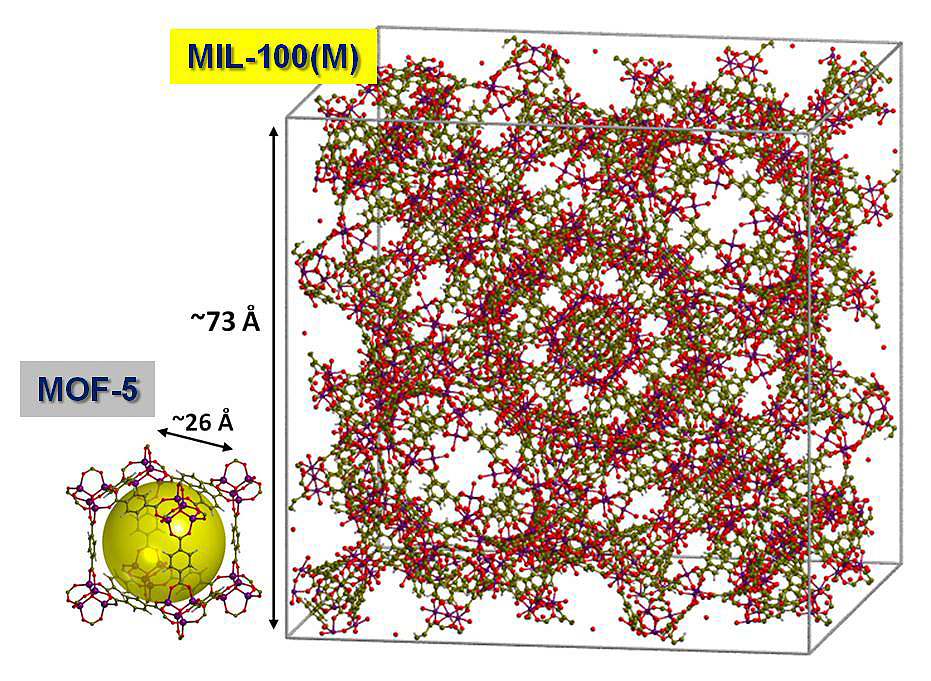
Figure 1: The comparison between the crystallographic unit cells of the giant MIL-100 (10,880 atoms) and MOF-5 (424 atoms) highlights the complexity of MIL-100 with respect to MOF-5 which is considered the archetype of MOFs. © Theoretical Chemistry Group – Department of Chemistry, University of Torino
MIL-100, with thousands of atoms in the unit cell (2,788 atoms in the primitive cell that is the one adopted for calculation), represents a tremendous challenge for current ab initio modelling. The use of Tier-0 computer resources provided by the Partnership for Advanced Computing in Europe (PRACE), such as HPC system SuperMUC of the Leibniz Supercomputing Centre in Garching near Munich, was essential to tackle this challenging problem. The periodic ab initio code CRYSTAL was used for the calculations (ab initio hybrid HF/DFT functionals augmented with a London-type correction).
Among the hottest topics in MOFs research in recent years has been the carbon dioxide capture. MIL-100 is characterized by the presence of open metal sites (OMS) exposed at the inner surface of the pores (see Figure 2) that greatly enhance its adsorptive properties. The adsorption of CO2 was then investigated for the isoreticular family of MIL-100 with different metals, namely: Al, Sc, Cr and Fe. Computed results were in qualitatively good agreement with experimental data thus confirming that the interaction of CO2 decreases along the series: Cr > Sc > Fe > Al. In contrast with experiment, results for different OMS showed that adsorption sites are rather homogeneous. This could be explained by the presence of defective sites in the real MOF structure.
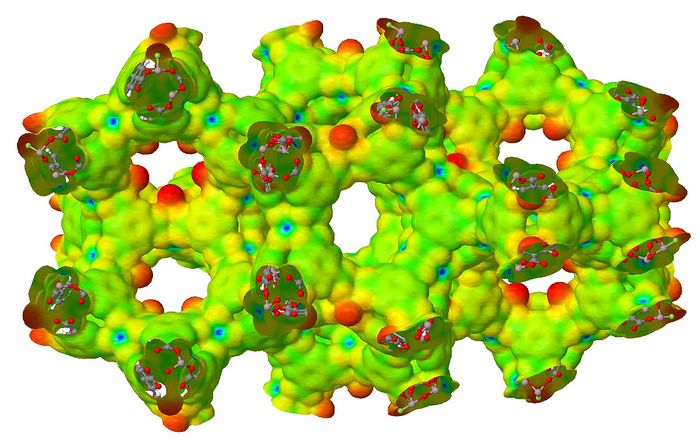
Figure 2. Electrostatic potential of MIL-100(Al) mapped on top of a charge density isosurface (0.003 e). Open metal sites are clearly visible as blue (positively charged) spots. © Theoretical Chemistry Group – Department of Chemistry, University of Torino
There is also a considerable interest in MIL-100 for its application in drug delivery. Exploratory calculations were also carried out for the anti-tumoral busulfan, widely used in chemotherapy regimes for leukaemias, to better understand the key forces acting between the drug and MIL-100(Fe).
This project was made possible by PRACE (Partnership for Advanced Computing in Europe) granting computing time on HPC system SuperMUC of LRZ.
Project team and scientific contact
Dr. Bartolomeo Civalleri (PI), Dr. Elisa Albanese, Dr. Maddalena D’Amore, Prof. Roberto Orlando
Theoretical Chemistry Group – Department of Chemistry, University of Torino
Via Giuria 7, I-10125 Torino (Italy)
e-mail: bartolomeo.civalleri@unito.it
LRZ project ID: pr85qu
March 2015