
MATERIALS SCIENCE AND CHEMISTRY
Capture and Release of CO2 in Monoethanolamine Aqueous Solutions: New Insights from First-Principles Reaction Dynamics
Principal Investigator:
Wanda Andreoni
Affiliation:
Institute of Theoretical Physics, Ecole Polytechnique Federale de Lausanne (Switzerland)
Local Project ID:
PRA069
HPC Platform used:
JUQUEEN of JSC
Date published:
The disastrous impact of anthropogenic carbon dioxide (CO2) emissions on the environment is very well known. The most mature technology for post-combustion CO2 capture, currently in use in the chemical industry, exploits a cyclic process, in which CO2 is selectively and reversibly absorbed in an amine (aqueous) solution. However, the operating costs are still too high to allow for large-scale implementation. A large empirical effort is ongoing worldwide primarily to reduce the high energy penalty required for amine regeneration and to increase the rate of CO2 absorption into the solvent.
There is a strong need for a quantitative characterization of the chemical reactions in all these processes and for a fundamental understanding of the microscopic mechanisms involved. Computer molecular simulations have the potential to assist scientists in the selection and optimization of the best engineering solutions by providing new insights on the relevant chemical processes and, at least in part, by delivering the quantitative information needed.
Scientists from the Ecole Plytechnique Federale de Lausanne in Switzerland leveraged the HPC capabilities of JSC system JUQUEEN to investigate the reference amine solution in industrial applications (as composition and concentration) with the aim of identifying the physico-chemical factors influencing the thermodynamics and kinetics of the key processes for the capture and release of carbon dioxide. The research benefited from frontier simulation methods and advanced software. In particular, it exploited the power of ab initio molecular dynamics, aided by accelerated sampling for rare events as provided by metadynamics techniques, and the calculations relied on the CPMD code that has the highest performance for this type of calculations on Blue Gene architectures.
The results achieved by Prof. Andreoni and her team were published in two journals:
(1) Journal of Physical Chemistry Letters
Abstract: Chemical absorption in amine aqueous solutions is a widespread technology for postcombustion carbon capture, and a large effort is ongoing to improve their performance. Characterization of the “reactant” and “product” solutions at the microscopic level is highly desirable for process optimization. Recently X-ray scattering experiments and “in situ” infrared spectroscopy have been applied to this aim, but a complete and convincing interpretation is missing. We present large-scale ab initio molecular dynamics simulations of monoethanolamine solutions at experimental concentration and temperature and analyze how structural and vibrational properties change after carbamate formation. An exhaustive account of the experimental data is obtained. Fingerprints of the reaction products and specific interactions are unravelled. Hydration effects are specific to each component of the solution and are essential for a correct assignment of the experimental data. Full report.
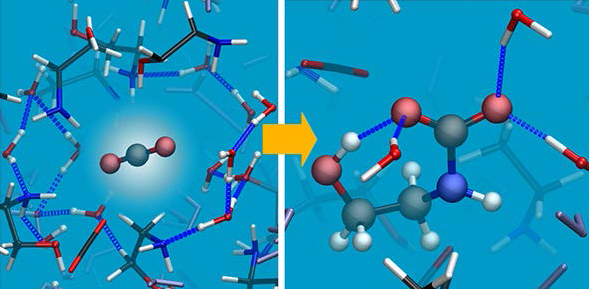
(2) Journal of Chemical Theory and Computation
Abstract: Aqueous monoethanolamine (MEA) solution is commonly used for postcombustion carbon capture via chemical absorption. Extensive research has been carried out to characterize both uptake and release of carbon dioxide (CO2), with the aim of improving process performance. However, an intensive research is still needed on fundamental aspects of the key chemical reactions, to achieve a comprehensive understanding of the cyclic process at the microscopic level and a quantitative assessment. We present several ab initio simulations of MEA solutions at a concentration of 30 wt % - the current standard in the industry - and study the dynamics of key multistep chemical reactions, using the metadynamics technique. Pathways for the entire cycle are investigated and characterized in terms of related free-energy and enthalpy barriers, and of the accompanying variations in both structural and electronic properties. The results of this study lead us to propose, among competing processes, an unforeseen scenario in which the zwitterion acts as an intermediate not only of CO2 uptake, in the form of carbamate, but also of its release. Rate-limiting steps are the formation of the zwitterion for the former and MEAH+ deprotonation for the latter. Water is shown to play a multifaceted role, which is crucial in determining the development and the energetics of each step of the reactions. The level of comprehension here achieved for MEA should help defining a strategy for solvent optimization.

The project was made possible through the Partnership for Advanced Computing in Europe (PRACE) granting computing time on HPC system JUQUEEN of JSC.
Scientific contact
Prof. Wanda Andreoni
Ecole Polytechnique Federale de Lausanne
Institute of Theoretical Physics
EPFL SB C3PN
PH H2 482 (Bâtiment PH)
Station 3
CH-1015 Lausanne/Switzerland
e-mail: wanda.andreoni [@] epfl.ch
JSC project ID: pra069
August 2015