
LIFE SCIENCES
Advanced Sampling of Peptide and Protein Association and Protein Conformational Transitions
Principal Investigator:
Martin Zacharias
Affiliation:
Department of Physics, Technical University Munich
Local Project ID:
pr27za
HPC Platform used:
SuperMUC and SuperMUC-NG of LRZ
Date published:
Introduction
Atomistic Molecular Dynamics (MD) simulations are widely used to study the dynamics of proteins and peptides and the association of biomolecules. However, the simulation time scales that can be reached are often still too short to reach biologically relevant conformational changes or to follow association processes.
We have developed and improved replica-exchange based advanced sampling methods to specifically tackle large scale conformational changes in biological macromolecules and to enhance the sampling of binding events. The techniques are based on the Hamiltonian replica exchange method. In several parallel running simulations specific biasing potentials are added to the force field (Hamiltonian) of the system. The biasing potential promotes transition in a collective degree of freedom or allows for more rapid association/dissociation of biomolecular complexes.
Due to possible exchanges between replica simulations including a reference system under the control of the original force field it is possible to specifically accelerate conformational transitions or binding events of interest. We have applied these techniques to study the 90kDa heat-shock protein (Hsp90) which is an essential molecular chaperon protein that plays a vital role in the folding process of several client proteins [1]. It is found in bacteria as well as eukaryotes and is essential for cell viability and plays a pivotal role in many signaling and regulation pathways. In its active conformation it forms a homodimer and its chaperone activity depends on ATP binding and hydrolysis. During its work cycle Hsp90 can adopt different global conformations covering a range of tightly bound closed structures up to widely open conformations [2,3].
In addition, we developed an efficient REMD method to accelerate the simulation of protein-protein and protein-peptide binding [4]. The approach can be used as efficient approach for predicting biomolecular complex formation. The approaches make efficient use of the SuperMUC parallel computer facilities.
Results and Methods
Hamiltonian replica exchange simulations on yeast Hsp90 middle and C-terminal dimer
In order to better understand the domain dynamics comparative Molecular Dynamics (MD) simulations of a substructure
of Hsp90, the dimer formed by the middle (M) and C-terminal domain (C), were performed. Since this MC dimer lacks the
ATP-binding N-domain it allows studying global motions decoupled from ATP binding and hydrolysis. Conventional (c)MD simulations starting from several different closed and open conformations resulted in only limited sampling of global motions. However, the application of a Hamiltonian Replica exchange (HREMD) method based on the addition of a biasing potential extracted from a coarse-grained elastic network (ENM) description of the system allowed much broader sampling of domain motions than the cMD simulations (termed ENM coupled REMD). The approach forms an effective multiscale methodology in which directions of large scale global conformational transitions (extracted from a low resolution technique) can guide and enhanced the high-resolution sampling of the multidomain structure [3] (Fig. 1).
With this multiscale approach it was possible to extract the main directions of global motions, the energy landscape and to elucidate the mechanism of the global structural transitions of the MC dimer [3]. Future applications to Hsp90 with the N-domains could be useful to elucidate global motions in the full Hsp90 molecule.
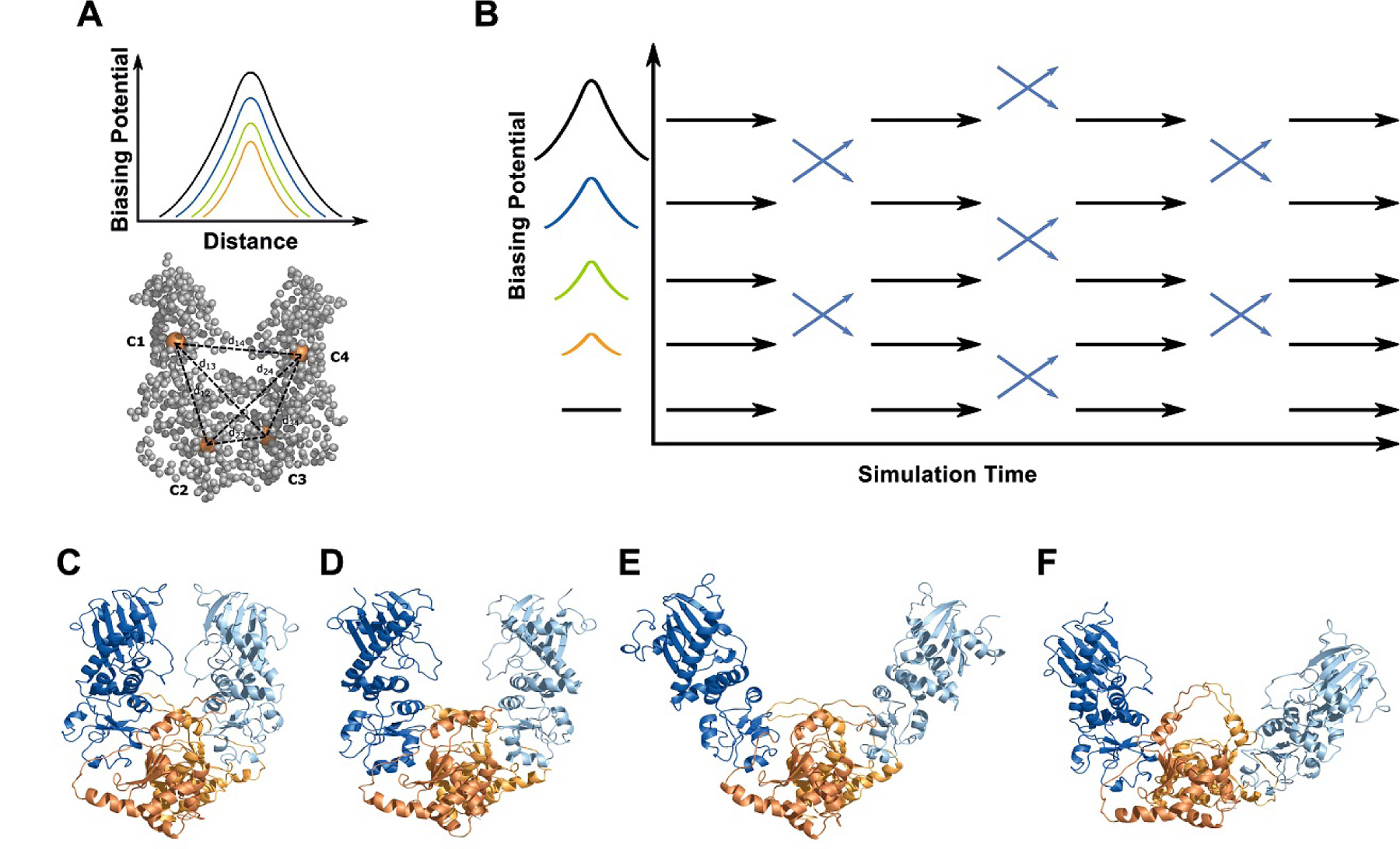
Figure 1: ENM coupled REMD simulations on a truncated yeast Hsp90 (dimer of M- and C-domains). (A) For the ENM-coupled REMD the center-of-mass of M- and C-domains (orange spheres, C1C4) served as the 4 centers to design a biasing potential (BP) in the replica runs. It acts along the distances between the 4 centers (dashed lines). The BP increases along the replicas (B) and the width is based on the distance fluctuation derived from the ENM calculations. (B) Illustration of ENM coupled REMD. (CF) Snapshots of global domain motion during the simulations (M: blue, C: orange). © TUM
Studying protein-protein and protein-peptide binding
In the addition to studying global protein motions, we developed a H-REMD based method to systematically investigate the formation of protein-protein and protein-peptide complexes and the refinement of predicted complexes. In each replica it includes different levels of a repulsive biasing between partners. The bias acts only on intermolecular interactions based on an increase in effective pairwise van der Waals radii (repulsive scaling (RS)-REMD) without affecting interactions within each protein or with the solvent (Fig. 2).

Figure 2: (left panels) Illustration of the biased van der Waals interaction along replicas runs during the RS-REMD approach. (right panels) Broad sampling of peptide (red) placements in highest replica around whole receptor (blue) and in intermediate replica as well as narrow sampling around the native binding site in the reference replica. The peptide start placement is in light blue. © TUM
For a set of 5 protein test cases (out of 6) the RS-REMD technique allowed the sampling of near-native complex structures even when starting from the opposite site with respect to the native binding site for one partner. The method allowed also efficient refinement of pre-docked protein-protein and protein-peptide complexes [4,5]. Even in case of relatively large protein-protein complexes the method allows the rapid sampling of near-native complex geometries. This was also possible when starting from the worst possible initial placement on the opposite side (with respect to the native binding site) of the surface of a partner protein (Fig. 3).

Figure 3: (upper panel) Snapshots of sampled starting, intermediate and near-native complexes (green: receptor; light grey: ligand protein; dark grey: native bound ligand) during a RS-REMD simulation (approximate simulation stage and sampling times are indicated). (lower panel) Ligand protein sampling density on receptor surface (light grey dots) in the first third, second third and final part of the simulation. © TUM
We have recently further extended the methodology such that a simultaneous refinement and free energy type of scoring of docked complexes is possible [5]. It is based on the observation that the technique samples both near-natively bound structures and dissociated states of a given protein-protein or protein-peptide complex along the replicas. The extensive overlap of sampled states in the replica simulations allows one to extract associated free energy differences and ultimately a free energy estimate of binding.
Apart from of identifying near-native binding geometries and estimating free energies of binding the methodology is also useful to characterize the full energy landscape for protein-protein binding on the surface of each partner protein and to identify transiently bound states as well as pathways from a non-specifically bound configuration towards the native complex geometry. It is very demanding but efficient on SuperMUC, and a step forward to a realistic predictionof protein-protein and protein-peptide interactions. An extension for refinement and free energy evaluation of docked drugreceptor complexes is in progress.
Ongoing Research / Outlook
Understanding the mechanism of how peptides or proteins bind to other partner molecules is of fundamental importance for basically all biological processes. With our new RS-REMD method we are in position to rapidly sample protein surfaces for potential binding sites under realistic solvation conditions and including full flexibility of binding partners. The new techniques allow a simultaneous refinement of potential complexes and at the same time a scoring of the complexes based on a free energy estimate. The approach for enhanced global domain motions is currently applied to multiprotein complexes involved in the process of RNA interference that is of major importance for regulatory processes of the cell. Due to the large size of these systems systematic simulations are only possible by using SuperMUC.
References and Links
[1] Pratt WB, Toft DO, Exp Biol Med 228 (2003) 111–33.
[2] Hellenkamp B, Wortmann P, Kandzia F, Zacharias M, Hugel T., Nat Methods 14 (2016) 174–180.
[3] Kandzia F, Ostermeir K, Zacharias M., Front Mol Biosci. 6 (2019) 93.
[4] Siebenmorgen T, Engelhard M, Zacharias M, J Comp. Chem. 10 (2020) 112.
[5] Siebenmorgen T, Zacharias M., J Chem Inf Model 60 (2020) 55525562
Research Team
Jonathan Coles1, Danial Pourjafar Dehkordi1, Julian Hartmann1, Manuel Hitzenberger1, Florian Kandzia1, Aliaksei Krukau2, Korbinian Liebl1, Gerald Mathias2, Asman Nayis1, Maria Reif1, Till Siebenmorgen1, Paul Westphälinger1, Martin Zacharias1 (PI)
1Technical University of Munich, Department of Physics (T38), Chair of Biomolecular Dynamics
2Leibniz Supercomputing Centre, Garching
Scientific Contact
Prof. Dr. Martin Zacharias
Technical University of Munich
Department of Physics (T38)
James-Franck-Str. 1, D-85748 Garching (Germany)
e-mail: zacharias [at] tum.de
NOTE: This report was first published in the book "High Performance Computing in Science and Engineering – Garching/Munich 2020 (2021)" (ISBN 978-3-9816675-4-7)
Local project ID: pr27za
November 2021