
LIFE SCIENCES
Scalable Computational Molecular Evolution Software & Data Analyses
Principal Investigator:
Alexandros Stamatakis
Affiliation:
Heidelberg Institute for Theoretical Studies (Germany)
Local Project ID:
pr58te
HPC Platform used:
SuperMUC of LRZ
Date published:
The field of phylogenetics reconstructs the evolutionary relationships among species based on DNA data. Substantial DNA sequencing technology advancements now generate a data avalanche. This allows using entire genomes of a large number of species for reconstructing phylogenetic trees. Statistical reconstruction approaches are widely used, but also highly compute-intensive. Researchers substantially improved the scalability and efficiency of two such statistical open-source tools on SuperMUC. In addition, they analysed several empirical large-scale datasets in collaboration with biologists.
Introduction
The field of phylogenetic tree reconstruction strives to infer the evolutionary relationships among a set of organisms (species, frequently also denoted as taxa) based on molecular sequence data. Recent advancements in sequencing technology, in particular the emergence of so-called next generation sequencers, have generated an avalanche of sequence data, that now makes it possible to use whole transcriptomes and even genomes of a large number of species for tree reconstruction.
Likelihood-based approaches (Maximum Likelihood and Bayesian Inference) represent an accurate and widely used, but at the same time also highly compute-intensive approach for reconstructing phylogenetic trees. In 2017 and 2018 we were able to substantially improve the scalability and efficiency of two Maximum Likelihood based tools for tree reconstruction and phylogeny-aware identification of anonymous molecular sequence data on SuperMUC.
In addition, we analyzed several empirical large-scale datasets in collaboration with biologists.
Scalable Software
A key focus of our lab is on developing methods in conjunction with large-scale empirical data analyses. In 2017 there has been substantial progress in developing and releasing novel, scalable open-source codes for phylogenetic inference. Our new tools rely on an open-source library for efficient phylogenetic likelihood calculations that is available as open source code under AGPLv3 (https://github.com/xflouris/libpll-2).
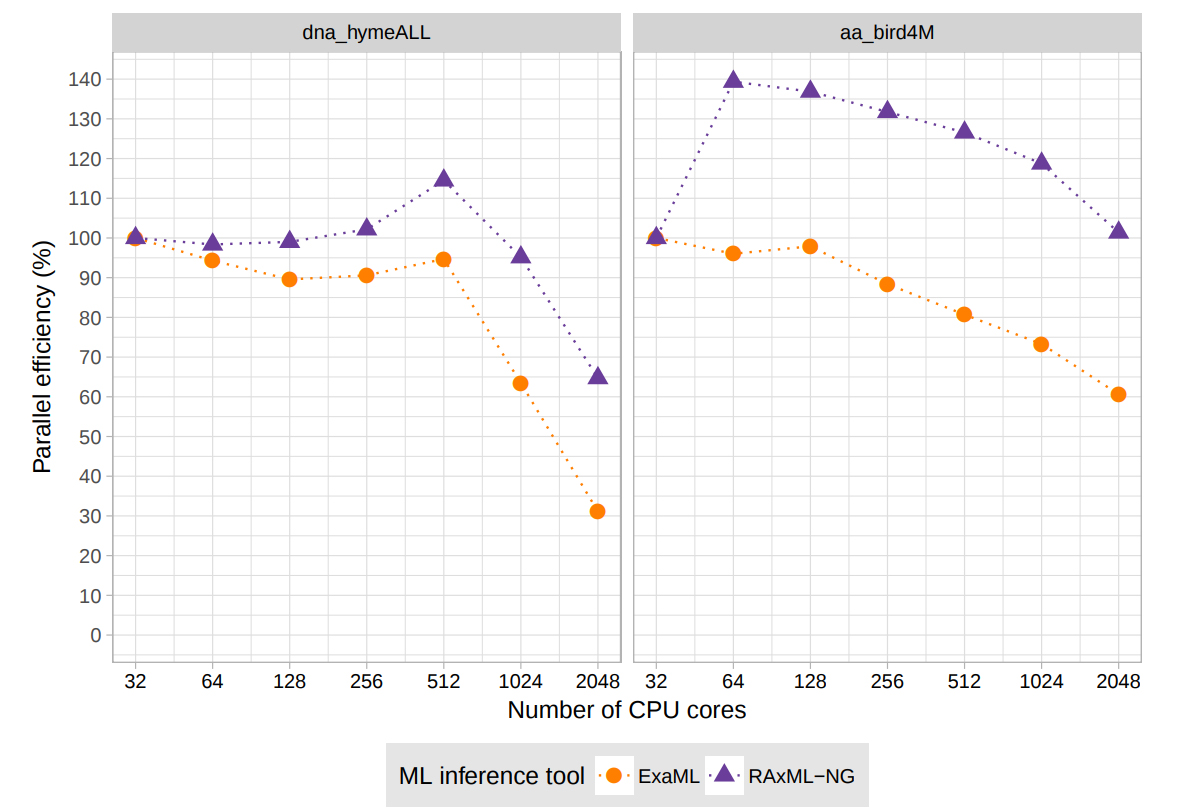
Figure 1. Super-linear speedups of the hybrid MPIPThreads version of RAxML-NG versus ExaML on large scale DNA (left) and amino acid datasets (right).
Copyright: HITS (Germany)RAxML-NG: In 2017, we released the complete re-design of our flagship tool for phylogenetic inference RAxML (over 20,000 citations on the four main papers, Google Scholar, accessed March 2018) as open source code under AGPLv3 (available at https://github.com/amkozlov/raxml-ng). RAML-NG has substantially superior sequential as well as parallel performance compared to RAxML and also compared to our previous dedicated tool for supercomputers (ExaML, see below). RAxML-NG integrates all optimizations from RAxML as well as ExaML and scales from the laptop to the supercomputer. In addition, we have designed a highly efficient hybrid parallelization that achieves spectacular super-linear speedups (up to 140%) due to increased cache efficiency. In Figure 1 we show a parallel efficiency comparison between RAxML-NG and ExaML on two large-scale DNA and amino acid datasets. Note that, that phylogenetic likelihood calculations are predominantly memory bandwidth bound.
EPA-NG: In 2017, we also released the complete re-design of our Evolutionary Placement Algorithm (EPA) as open-source code under AGPLv3 (available at https://github.com/Pbdas/epa-ng).
The EPA places anonymous sequences as obtained from metagenetics studies onto a given reference phylogeny using the Maximum Likelihood criterion. As a data analysis of protists living in neotropical forest soils revealed (mentioned in our previous report, published in Nature Ecology & Evolution in early 2017; also see, e.g., press coverage https://insidehpc.com/2017/03/supermuc-helps-discover-new-species-critical-rainforest-ecosystems/) our previous implementation had reached its performance limits as the number of molecular sequences produced by such studies steadily increases. The new version is between 3.5 to 370 times faster than our previous implementation (depending on heuristic parameter settings) and also 30 times faster than a competing tool for the same purpose called pplacer. In addition, we have also designed a novel parallel version of the tool that exhibits good parallel strong scaling efficiency (see Figure 2).
While the papers describing RAxML-NG and EPA-NG have not been submitted yet, we believe that both are likely to become high impact papers.
Except for the tools presented here, we have also developed and released a new tool for phylogenetic model testing and continued work on improving load balance of phylogenetic likelihood calculations via appropriate data distribution algorithms [5].

Figure 2. Strong parallel scaling efficiency of EPA-NG for placing 10 million, 100 million, and 1 billion molecular sequences into a phylogenetic reference tree on up to 2048 cores.
Copyright: HITS (Germany)Scalable Data Analyses
In 2016 and 2017, we still used our previous dedicated supercomputer codes – ExaML and ExaBayes – to conduct several large-scale phylogenomic analyses in the context of the ongoing 1KITE project (www.1kite.org). In particular, our work shed new light on the evolutionary history of a large group of insects that includes wasps, bees, ants, and sawflies (order Hymenoptera). This group exhibits several interesting evolutionary transitions, for instance, from plant-feeding to predation and parasitism (and back to pollen-collecting in bees), or from solitary to eusocial lifestyle.
We inferred a phylogeny of 173 Hymenoptera species using 3,256 protein-coding genes (>1,500,000 alignment columns). For thoroughly analyzing this large dataset, we used ~650,000 CPU-hours in total, while each individual run typically used 640 (ExaML) up to 1792 (ExaBayes) cores. Notably, we performed one of the largest Bayesian phylogenetic analysis to date and set new standards for what is feasible with current software and hardware in this area. The resulting phylogenetic tree is depicted in Figure 3.

Figure 3. Phylogenetic tree of the Hymenoptera.
Copyright: Current BiologyTwo smaller studies that focused on vespid wasps [3] and chalcid wasps [4] have been published in Molecular Phylogeny and Evolution. Among other findings, they confirmed that several important traits such as eusociality or the ability to jump have evolved multiple times independently in different wasp lineages.
Finally, we executed analogous phylogenomic analysis for three further insect subgroups: Syrphoidea (hoverfiles), Apoidea (wasps and bees), and Paraneoptera (lice and thrips). The corresponding papers have either been accepted (Syrphoidea in Systematic Entomology) or are under review.
On-going Research / Outlook
With our novel efficient parallel software now in place (RAxML-NG and EPA-NG), we are ready to conduct further challenging large-scale phylogenetic analyses on SuperMUC and SuperMUC-NG. The key goal for 2018 is to analyze the final and extremely large insect dataset in the framework of the 1KITE project. This dataset contains roughly 1000 genes from about 1300 species. This dataset is particularly challenging as it contains, both, a large number of genes and a huge number of taxa. Note that, previous analyses of insect datasets on SuperMUC only contained between 100 – 200 species.
Another key challenge is to further optimize the I/O efficiency of EPA-NG as it constantly reads molecular sequence data from file and also generates large result files (current measured I/O throughput is 5Gbit/s).
Research Team
Pierre Barbera, Alexey Kozlov, Alexandros Stamatakis (PI)
References and Links
[1] lab web-site: www.exelixis-lab.org
[2] R. S. Peters, L. Krogmann, C. Mayer, A. Donath, S. Gunkel, K. Meusemann, A. Kozlov, L. Podsiadlowski, M. Petersen, R. Lanfear, P. A. Diez, J. Heraty, K. M. Kjer, S. Klopfstein, R. Meier, C. Polidori, T. Schmitt, S. Liu, X. Zhou, T. Wappler, J. Rust, B. Misof, O. Niehuis. “Evolutionary History of the Hymenoptera”. Current Biology 27(7): 1013-1018, 2017.
[3] S. Bank, M. Sann, C. Mayer, K. Meusemann, A. Donath, L. Podsiadlowski, A. Kozlov, M. Petersen, L. Krogmann, R. Meier, P. Rosa, T. Schmitt, M. Wurdack, S. Liu, X. Zhou, B. Misof, R. S. Peters, O. Niehuis. “Transcriptome and target DNA enrichment sequence data provide new insights into the phylogeny of vespid wasps (Hymenoptera: Aculeata: Vespidae)”. Molecular Phylogenetics and Evolution 116: 213-226, 2017.
[4] R. S. Peters, O. Niehuis, S. Gunkel, M. Bläser, C. Mayer, L. Podsiadlowski, A. Kozlov, A. Donath, S. van Noort, S. Liu, X. Zhou, B. Misof, J. Heraty, L. Krogmann. “Transcriptome sequence-based phylogeny of chalcidoid wasps (Hymenoptera: Chalcidoidea) reveals a history of rapid radiations, convergence, and evolutionary success”. Molecular Phylogenetics and Evolution, 120: 286-296, 2018.
[5] B. Morel, T. Flouri, A. Stamatakis. "A novel heuristic for data distribution in massively parallel phylogenetic inference using site repeats". IEEE HPCC17, 2017.
Scientific Contact
Prof. Dr. Alexandros Stamatakis
Scientific Computing Group (SCO), Heidelberg Institute for Theoretical Studies
HITS gGmbH, Schloss-Wolfsbrunnenweg 35, D-69118 Heidelberg (Germany)
Email: alexandros.stamatakis [@] h-its.org
NOTE: This report was first published in the book "High Performance Computing in Science and Engineering – Garching/Munich 2018".
LRZ Project ID: pr58te
March 2019