
LIFE SCIENCES
AVOCADO - AdVanced fOrmulation using Coarse grAineD mOdelling
Principal Investigator:
Dr. Benjamin Winkeljann
Affiliation:
Ludwig Maximilians Universität München, Munich, Germany
Local Project ID:
pn25ma
HPC Platform used:
SuperMUC-NG PH-1 CPU at LRZ
Date published:
Introduction
Biologics offer significant therapeutic advancements, but their complex nature poses formulation challenges due to their sensitivity to environmental factors. Molecular dynamics (MD) simulations can accelerate formulation development by investigating biophysical behavior and guiding the design of stable and effective formulations. However, MD simulations face limitations in computation-al resources and force field accuracy. This report details the use of supercomputing capabilities for large-scale MD simulations, exploring various formulation parameters using coarse-grained (CG) models. Experimental data was used to cross-validate simulation results, ensuring the reliability of insights and demonstrating the potential to streamline the development of optimized biologic formulations.
Results and Methods
Subproject 1: Investigating nucleic acid delivery and interaction with polycationic vectors.
Polymeric nanoparticles for RNA drug delivery commonly range in size from around 10 to 200 nm and self-assemble in an incubation time of minutes to hours. To retrieve comprehensive insights on the structural organization of those particles via MD simulations, sufficient simulation box sizes and extended time spans are prerequisite. The computational power to assess such systems was provided by the SuperMUC-NG.
This setup enabled a screening for influencing factors on particle structure: Firstly, we identified the hydrophilicity (represented by the percentage of lipophilic moieties (% OA)) of our polymers as the main parameter for particle morphology (Figure 1). Secondly, changes in ionic strength and buffer pH were recognized to influence
polymer self-interactions and aggregation tendencies of the particles. Identification of these factors allows correlation to wet lab results and trends seen in in vitro experiments. Additionally, it will lead to optimization of simulation protocols to efficiently characterize further particle systems.
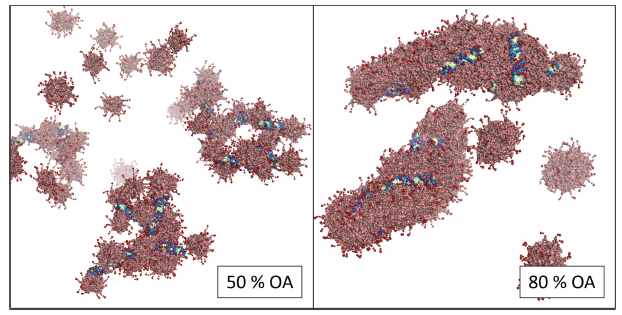
Figure 1: Influence of the lipophilicity of the polymers (%OA) on nanoparticle morphology.
Subproject 2: Protein Aggregation at Moving Interfaces.
We developed a CG MD setup designed to replicate the proposed mechanism of protein aggregation at moving interfaces such as tubing during transfer and filling opera-tions. Given the necessity to simulate a high number of large systems for statistically significant results, employ-ing job farming was essential for the success of this project. The majority of these simulations were non-equilibrium MD simulations, demanding increased computational power. Our simulation outcomes correlated with experimental data on subvisible particles and turbidity, thereby validating our model. Using this model, we were able to gain mechanistic insights. For instance, we found that compression velocity does not affect the aggregation behavior of preformed protein films, but rather influences their regeneration. With the possibility of studying the impact of different variables upon compression and dilation at the interface on a molecular level, our model contributes to the understanding of the mechanisms of protein aggregation at moving interfaces.
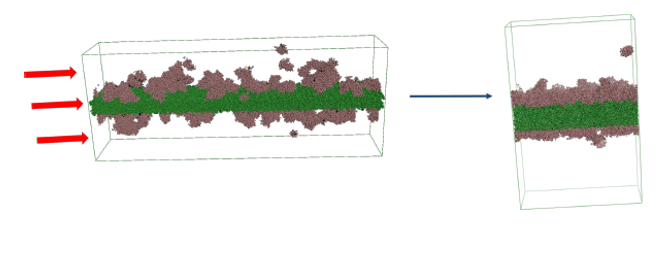
Figure 2: MD setup for protein aggregation at interfaces.
Subproject 3: Rational design of the antibody ion cloud targeting protein-self-interactions.
Although CG-MD simulations have been shown to provide valuable insights for investigating nanoscale phenomena, commonly used CG force fields, such as Martini 2, often suffer from overestimation of protein-protein interactions. This inaccuracy limits their ability to reliably predict important solution properties (e.g., B22 value, viscosity, aggregation propensity), which are critical for computer-aided drug and formulation design. While the newer Martini 3 force field improves modeling, it still exhibits discrepancies in protein-protein interactions, highlighting the need for further refinement to better match experimental data. We developed a novel approach combining metadynamics and umbrella sampling with the Martini 3 force field to investigate protein-protein interactions. By calculating potentials of mean force across various proteins and salt concentrations, we identified systematic overestimations in the force field. To enable this systematic approach, a large number of simulations were necessary, which was enabled by a job farming approach on SuperMUC-NG.
Subproject 4: Simulation of true-sized polyethyleneimine (PEI) for the complexation of RNA drugs.
A CG model for PEI was developed within the Martini 3 forcefield, and MD simulations were executed to investigate the complexation between PEI and siRNA. A code was developed that allows for the generation of input files for MD simulations of PEI at its true size (several kilodalton) and variable degrees of branching. We further proposed a methodology for systematically mapping large molecular structures through fragmentation. Electrostatic interactions were identified as central to the process, with molecular weight and branching of PEI playing pivotal roles. The importance of a combined computational and experimental approach in understanding these complex systems is highlighted. On the computational side, we employed metadynamics, much like Subproject 2 and 3. This approach is computationally very intensive, requiring large numbers of cores to calculate the complex free energy landscapes along predefined collective variables.
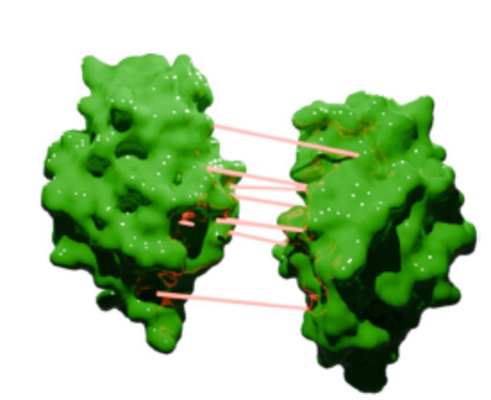
Figure 3: Visualization of the molecular interaction between two lysozyme molecules.
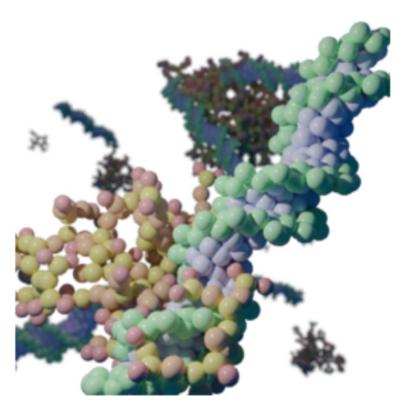
Figure 4: Complexation of siRNA with PEI molecules.
Ongoing Research / Outlook
While our initial simulations on supercomputing systems have yielded promising results that were validated by experimental data, MD's potential is limited by computational constraints and force field accuracy. To fully explore the complexities of biologic formulations, more simulations covering longer timescales, diverse parameters, and refined coarse-grained models are essential. This demands a significant increase in computing resources and core hours to advance biologic formulation strategies and ultimately benefit patient outcomes.
References and Links
[1] Tim Sarter and Wolfgang Frieß, Molecular Pharmaceutics (2024). https://github.com/jonbind/closingthegap
[2] Jonas Binder, Joshua Winkeljann, Katharina Steinegger, Lara Trnovec, Daria Orekhova, Jonas Zähringer, Andreas Hörner, Valentin Fell, Philip Tinnefeld, Benjamin Winkeljann, Wolfgang Frieß, and Olivia M. Merkel, Molecular Pharmaceutics (2024). https://doi.org/10.1021/acs.molpharmaceut.3c00747