
LIFE SCIENCES
Enzyme Design by QM/MM Monte Carlo
Principal Investigator:
Ville R. I. Kaila
Affiliation:
Department of Chemistry, Technical University of Munich (Germany)
Local Project ID:
pr74ve
HPC Platform used:
SuperMUC of LRZ
Date published:
Introduction
We have developed a new method for computational protein design where we randomly mutate amino acids of enzymes using a Metropolis Monte Carlo (MC) procedure. The aim of the method is to identify substitutions that increase the catalytic activity, i.e., lower the reaction barrier for catalysis. We probe the catalytic activity by quantum mechanics/classical mechanics (QM/MM) calculations, which are important for accurately modeling chemical reactions [1].
Results and Methods
The QM/MM Monte Carlo (QM/MM MC) method has been tested using a computationally designed enzyme for a catalytic reaction step, here called Enzyme A. We wanted to predict mutations that would increase the efficiency of this enzyme. A related enzyme referred to as Enzyme B served as a reference to the predicted mutations. Enzyme B was obtained experimentally performing mutations on Enzyme A in order to improve its catalytic efficiency.
The QM/MM MC method follows a mutation-equilibrationevaluation scheme. In the first step, the initial structure of the enzyme undergoes a random mutation move. The mutations are performed using the molecular modeling software Visual Molecular Dynamics (VMD) [2]. The mutated protein structure is equilibrated by classical molecular dynamics (MD) simulations employing the software NAMD [3]. After the MD relaxation step, the reaction barrier is probed at quantum chemical level employing the program TURBOMOLE [4], by performing QM calculations on pre-optimized reactant and transition state (TS) structures of the substrates within the mutated and relaxed enzyme structure. Based on the calculated reaction barrier, the mutation move is either accepted or rejected by applying the Metropolis Monte Carlo algorithm, following the next mutation cycle. The method has been implemented using the programming language Python.
During the beginning of the Summer of Simulation 2016, the QM/MM MC method was implemented on the SuperMUC supercomputer. Using the parallelization framework Redisexec, N independent protein replicas, generated from the same starting structure, were simulated in parallel on the SuperMUC thin nodes, with each replica occupying one node. In these simulations, the QM calculations were performed in a sequential way. Scaling tests were performed for the QM/MM MC method within the parallelization framework Redisexec on the SuperMUC thin nodes with 16 CPUs each, subjecting each replica to five mutation moves, using one node per replica (Figure 1). Concluding from the scaling tests, 100 nodes were used within the parallelization framework Redisexec for the upcoming QM/MM MC calculations.
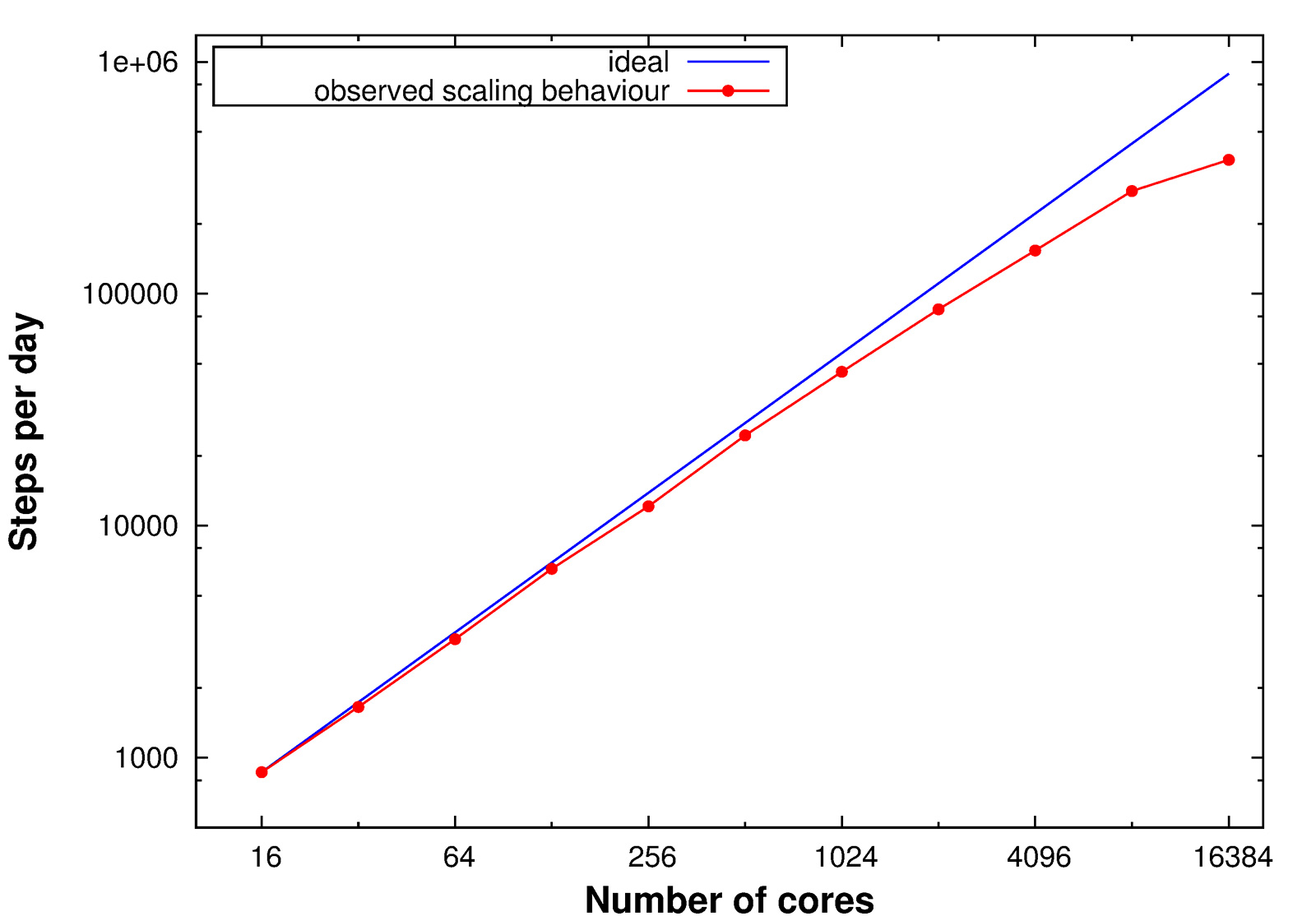
Figure 1: Scaling tests with five single mutations performed per node.
Copyright: Group for Computational Biocatalysis, Department of Chemistry, TUMIn order to reduce the time required for one mutation step, the implementation was modified to allow every replica to be calculated using two nodes instead of one. This way, the time needed for the MD relaxation step was reduced and both of the QM calculations were performed in parallel at the same time. All in all, the average time needed to perform one mutation step decreased from ~2 minutes to ~1.3 minutes. Analysis of the mutations performed during multiple mutation trajectories shows that residues close to the active site are mutated most frequently (Figure 2). The computationally sampled mutations were found to reduce the approximated barrier from ~10 kcal mol-1 to ~1 kcal mol-1.
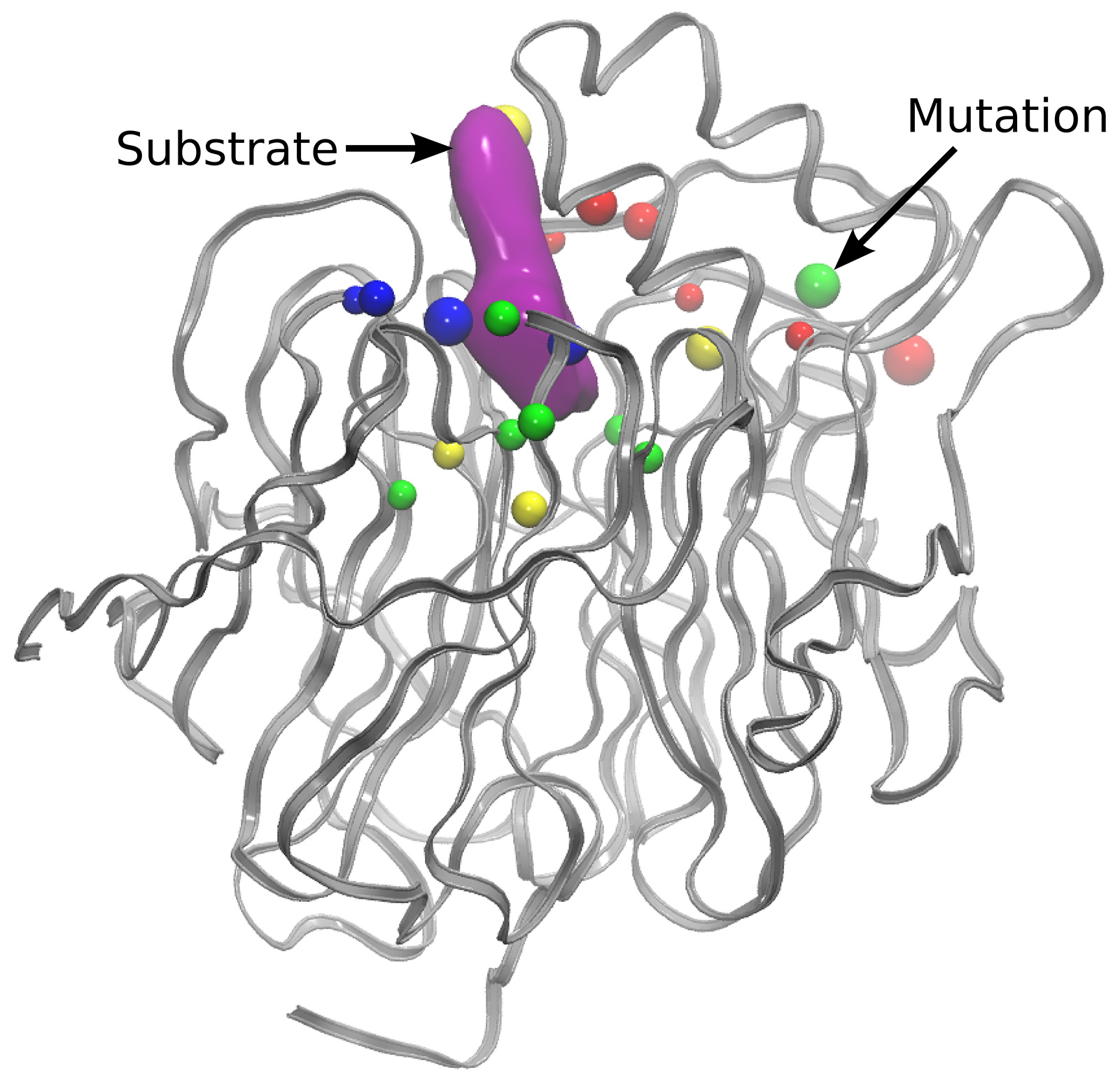
Figure 2: Frequently mutated residues of Enzyme A with their most frequent mutation type shown using the following color code: blue: positively charged, red: negatively charged, yellow: polar, green: non-polar.
Copyright: Group for Computational Biocatalysis, Department of Chemistry, TUMWe also computed free energy profiles for the reactions catalyzed by Enzyme A and Enzyme B, respectively, by employing the QM/MM umbrella sampling technique. To this end, 23 independent QM/MM MD simulations were performed for each of the enzymes, by subsequently restraining the conformations along the reaction coordinate, going from the reactant state to the product conformation. The 46 conformations were sampled for 1 picosecond each, employing the CHARMM/TURBOMOLE interface [5]. These calculations were performed using the Redisexec framework as well, using one node per conformation.
The resulting free energy profiles are qualitatively in agreement with the experimental findings as the calculated reaction barrier for Enzyme A is higher than for Enzyme B (Figure 3).
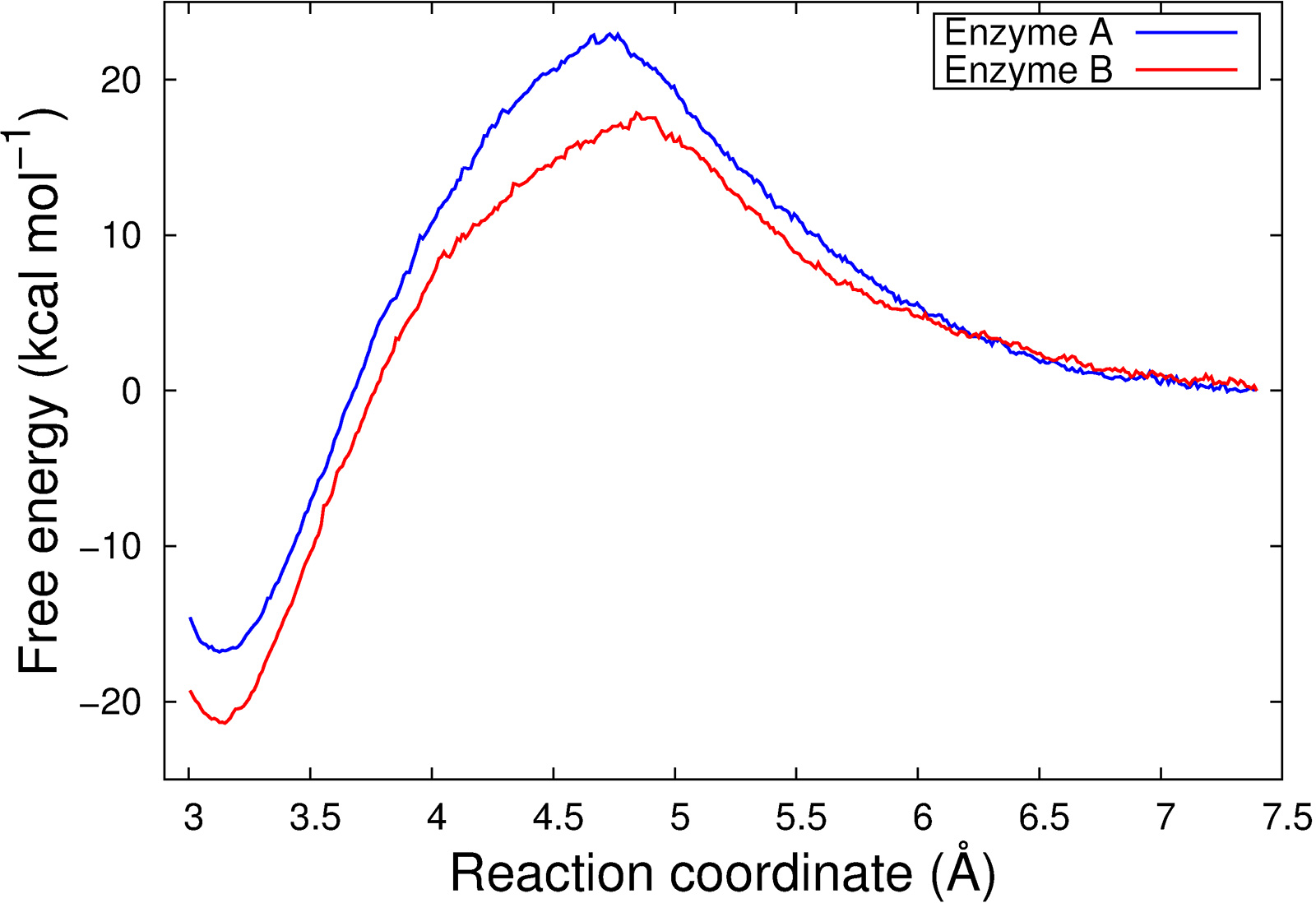
Figure 3: Free energy profiles for the reaction steps catalyzed by Enzyme A and Enzyme B.
Copyright: Group for Computational Biocatalysis, Department of Chemistry, TUMThe project consumed in total 6 million CPU hours. For every replica, ca. 2000 files were generated. Overall, ~1.5 TB of storage were needed, distributed over SCRATCH and PROJECT.
On-going Research / Outlook
SuperMUC provided us with the computing time we needed to test and apply our method. SuperMUC Phase 2 turned out to be most suitable for certain TURBOMOLE calculations. During the scaling tests for the QM/MM MC method on 1024 nodes, the quota in PROJECT was filled up with socalled “in-doubt blocks”, possibly produced by the TURBOMOLE calculations. In order to avoid incidents like this for the upcoming scaling tests and production runs, the temporary files produced by TURBOMOLE were removed after each mutation cycle, and the production runs were performed on the SCRATCH file system.
Running more than 100 replicas in parallel, the increased number of I/O operations seemed to slow down the overall performance of the framework. The catalytic activity of the mutated structures will be investigated further by both computational and experimental means. The umbrella sampling technique will be applied to the most promising mutated structures obtained from our QM/MM MC method, in order to verify that the reaction barrier of Enzyme A has been lowered.
References and Links
[1] https://www.compbio.ch.tum.de
[2] William Humphrey, Andrew Dalke, and Klaus Schulten. 1996. VMD: Visual Molecular Dynamics. J. Mol. Graphics 14, 1 (Feb. 1996), 33–38. DOI: https://doi.org/10.1016/0263-7855(96)00018-5
[3] James C. Phillips, Rosemary Braun, Wei Wang, James Gumbart, Emad Tajkhorshid, Elizabeth Villa, Christophe Chipot, Robert D. Skeel, Laxmikant Kalé, and Klaus Schulten. 2005. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 26, 16 (Dec. 2005), 1781–
1802. DOI: https://doi.org/10.1002/jcc.20289
[4] Reinhart Ahlrichs, Michael Bär, Marco Häser, Hans Horn, and Christoph Kölmel. 1989. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 162, 3 (Oct. 1989), 165–169. DOI: https://doi.org/10.1016/0009-2614(89)85118-8
[5] Saleh Riahi and Christopher N. Rowley. 2014. The CHARMM-TURBOMOLE Interface for Efficient and Accurate QM/MM Molecular Dynamics, Free Energies, and Excited State Properties. J. Comput. Chem. 35, 28 (Oct. 2014), 2076-2086. DOI: https://doi.org/10.1002/jcc.23716
Research Team
Prof. Dr. Ville R. I. Kaila (PI), Sophie L. Mader
Scientific Contact
Prof. Dr. Ville R. I. Kaila
Department of Chemistry
Technical University of Munich (TUM)
Lichtenbergstr. 4, D-85747 Garching (Germany)
e-mail: ville.kaila [@] ch.tum.de
NOTE: This report was first published in the book "High Performance Computing in Science and Engineering – Garching/Munich 2018".
LRZ project ID: pr74ve
October 2019