
LIFE SCIENCES
Replica Exchange Molecular Dynamics Simulation of the Switching Process in small GTPases
Principal Investigator:
Martin Zacharias
Affiliation:
Lehrstuhl für Molekulardynamik, Physik-Department T38, Technische Universität München (Germany)
Local Project ID:
pr74bi
HPC Platform used:
SuperMUC of LRZ
Date published:
Small GTPase protein molecules mediate cellular signaling events by transient binding to other proteins that in turn activate or deactivate processes in the cell. The signaling of GTPase proteins is mediated by switching between different active or inactive conformational states. Understanding the molecular details of these switching events is of great importance to understand cellular regulation and to design drug molecules to control cell functions. Using Molecular Dynamics advanced sampling techniques, the mechanism of conformational switching in the Rab8a-GTPase were investigated.
Introduction
Small GTPases are a class of protein-based switches that play a significant role in many intracellular signalling events by switching between active and inactive states [1]. Depending on the switch state, its two important regulatory regions, namely switch I and II, adopt different conformations. While in the active state both regions are highly conserved and ordered, upon inactivation they become disordered [2].
The conformation and flexibility of switch regions are crucial for recognising the physiological binding partners. It has been revealed that post-translational modifications (PTMs) - such as phosphorylation, phosphocholination, or adenylylation - of specific amino acid residues modulate their activity [3]. Rab, Rho/Cdc42 and Ras GTPase subfamilies are among the most frequently targeted ones.
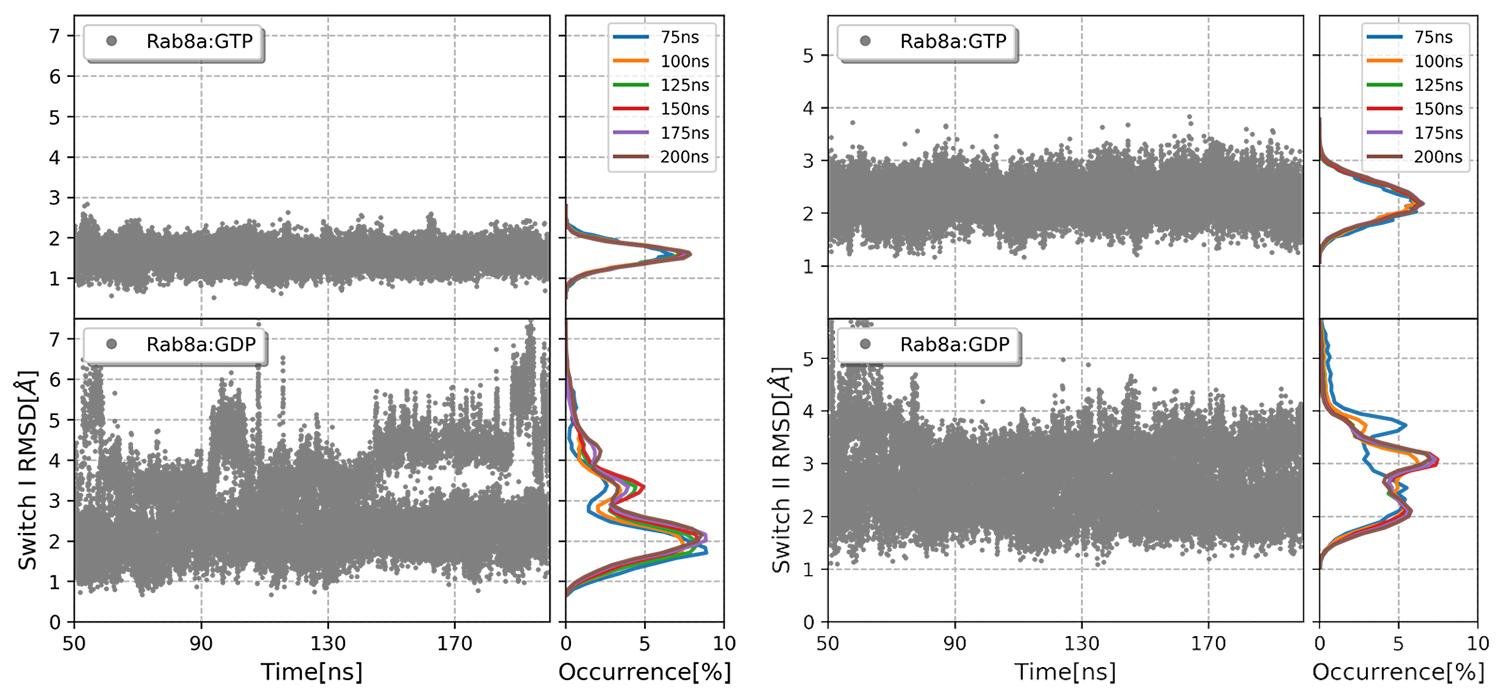
Figure 1: Root mean square deviation of switch I and switch II with respect to the active Rab GTPase structure.
Copyright: TU MünchenPhosphorylations by protein kinases inside or outside the switch regions also manipulate the activity of G-proteins, as observed in Rab8a where Ser111, not inside the switch regions, is phosphorylated. The conformational consequences of such modifications leads us to understand their biological impact.
The aim of this work, which is part of a project in collaboration with experimental groups within the framework of SFB1035 on control of protein function by conformational consequences of such modifications, is to understand how the switching mechanism is modulated through specific modifications by means of molecular dynamics (MD) and advanced sampling simulation techniques. Combination of MD and biased potential replica-exchange (BPRE) help us obtain an insight, in atomic details, on how PMTs interfere with GTPases switching mechanism.
Results and Methods
Our method relies on introducing a biased potential that promotes conformations with unfavourable backbone and side-chain dihedrals. The starting setup was composed of eight parallel MD simulations (also referred to as “replicas”) of the solvated protein-ligand complex. Along the replicas, the potential was increased to penalise low energy angles, namely the ones associated with α-helices and β-sheets. Based on a Metropolis criterion, every 1000 steps an exchange attempt between the neighbouring replicas was allowed or rejected. The idea is that if we have enough exchange attempts, at the point of convergence there will be an unbiased representation of the conformational space.
For simulating each complex, that is composed of 22,480 atoms, including water molecules, 448 cores were deployed. Overall, 7 million of the granted 8 million CPU-hours were used. The biasing potential was targeted on switch I (residues 30-42) and switch II (residues 64-80) of both ligand-bound complexes.
We started the simulations this time at 315K at which the behaviour of the two complexes became clearly distinguishable in the switch regions. RMSD values of switch I indicated a higher occurrence of larger deviations i.e. frequent exchange to the higher replicas, in Rab8a:GDP compared to Rab8a:GTP (Fig.1). Switch II illustrated clearly higher flexibility in Rab8a:GDP complex, with peak RMSD values at 2 and 3.8Å, whereas in GTP-bound Rab the deviations were limited to 2Å.

Figure 2: Distribution of the switch I (up) and II (down) domains RMSD of the heavy atoms, with respect to the initial position.
Copyright: TU MünchenThe outcome clusters of both complexes varied merely in the switch regions. In the GTP-bound Rab, the characteristic interactions between T40 and the γ phosphate and the Mg2+ ion remained intact, whereas in Rab8a:GDP, the absence of this bond facilitates switch II unfolding. Analysis of the switch II region revealed that although the helix was separated from the ligand binding pocket, it restrained the α-helical conformation.
In the next step, we used our BPRE model to investigate the effects of T72 and S111 phosphorylation on Rab8a structure. Comparing the RMSD of the switch regions showed that the phosphorylated Rabs, although bound to GDP, were stabilised in a conformation close to that of Rab8a:GTP complex (Fig.2).
This shift was more profound in switch II, where one of the phosphorylated residues is directly located (T72) and the other one is in remote interaction with (S111). Investigating the clusters obtained from the BPRE simulations showed that indeed there were new non-native polar contacts formed between the added phosphate groups and their neighbouring amino acid residues that stabilised the switch II region in its active conformation (Fig.3).

Figure 3: Most populated clusters obtained from BPRE simulation of T72 phosphorylated Rab8a:GDP (orange) overlaid on wild-type Rab8a:GDP:Rabin8 crystalline structure. New hydrogen bonds are formed between phosphorylated T72 and the neighbouring residues.
Copyright: TU MünchenComparing the binding free energies (Tab.1) indicated that the complex of Rabin8 and GDP-bound Rab8a is favoured to all other ensembles. The energy of Rabin8 binding to the wild-type Rab8a:GDP was favoured over pS111- Rab8a:GDP and pT72-Rab8a:GDP by about -5.71 and -49.16 kCal/mol, respectively. The drastic worsening in the latter case may be due to the position of T72 that is directly at the interface of Rab8a:Rabin8.

Table 1: Absolute (∆G) and relative (∆∆G) free energy differences of Rab:Rabin binding with respect to GDP bound complex in kCal/mol. The free energies were calculated using MM-PBSA package of Amber software from a 5ns production run.
Overall, using the BPRE model we were able to address two important implications of PTMs on Rab8a. First, the switch regions are stabilised in a closed form that is similar to the active GTP bound conformation, which obstructs switch domains opening necessary for the ligand exchange. Second, the interactions between the target residues and the interacting partner (Rabin8) are disturbed due to new hydrogen bonding competition. These two factors effectively impair Rab8a-Rabin8 interactions which results in a drastically reduced nucleotide exchange rate for phosphorylated Rab, which is in agreement with the experimental results[4].
On-going Research / Outlook
There are several other members of the Ras superfamily that are prone to modifications which will be investigated. Modifications that are of various types have to be examined by our method. Moreover, it has to be investigated wether with the presence of the binding partner, the above discussed conformational changes still take place. Following these objectives will help us have a clear understanding of the implications of an advanced sampling method in predicting the consequences of a chemical modification on Ras protein family.
References
1. Schwartz, Samantha L., et al. "Rab GTPases at a glance." Journal of cell science 120.22 (2007): 3905-3910.
2. Prakash, Priyanka, and Alemayehu A. Gorfe. "Lessons from computer simulations of Ras proteins in solution and in membrane." Biochimica et Biophysica Acta (BBA)-General Subjects 1830.11 (2013): 5211-5218.
3. Lai, Yu‐Chiang, et al. "Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1." The EMBO journal 34.22 (2015): 2840-2861.
4. Lai, Yu‐Chiang, et al. "Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1." The EMBO journal 34.22 (2015): 2840-2861.
Researchers
Danial Pourjafar-Dehkordi, Martin Zacharias (PI)
Scientific Contact:
Prof. Dr. Martin Zacharias
Lehrstuhl für Theoretische Biophysik (T38) - Molekulardynamik
Technische Universität München
James-Franck-Str. 1, D-85748 Garching (Germany)
e-mail: martin.zacharias[at]mytum.de
NOTE: This report was first published in the book "High Performance Computing in Science and Engineering – Garching/Munich 2018".
LRZ project ID: pr74bi
May 2019