
LIFE SCIENCES
Structure and Dynamics of Nascent Peptides in the Ribosome Exit Tunnel
Principal Investigator:
Helmut Grubmüller
Affiliation:
Max Planck Institute for Biophysical Chemistry, Göttingen (Germany)
Local Project ID:
pr62de
HPC Platform used:
SuperMUC of LRZ
Date published:
The ribosome is a complex molecular machine which plays an essential role in protein biosynthesis across all domains of life. Knowing its structural and mechanistic details may help to develop new medical treatments by controlling protein production or to understand the context of neurodegenerative diseases. Using molecular dynamics simulations this project studies how certain nascent peptides, similar to particular antibiotics, affect the transport of produced polypeptide chains through the exit tunnel rendering this process moreover an attractive target from a pharmacological perspective.
Introduction
The ribosome is a stochastic nanomachine responsible for protein synthesis in all cells (Fig 1). It is a large biomolecular complex of several ribonucleic acid (RNA) strands, and of a few dozen proteins. Ribosomes translate the genetic information from the messenger RNA (mRNA) into a sequence of amino acids. The process of translation is regulated by several factors and involves a cyclic binding of transfer RNAs (tRNAs) into three specific sites (labeled A, P and E). Ribosomes are highly conserved across all domains of life. Any malfunction of the biomachine notably affects the cellular life cycle. Apart from their fascinating nature and function, ribosomes represent one of the most important targets for antibiotic binding [1].

Figure 1: Anatomy of the bacterial ribosome (PDB: 5afi)
Copyright: MPI BPC, GöttingenAt the turn of the millennium, the atom-resolved structure of a prokaryotic ribosome was determined. Since then, a great quantity of structural information has been obtained from several species including Escherichia coli, Bacillus Thermophilus, yeast and humans. Ribosomes read mRNA at a decoding site on their small subunit (30S). The peptidyl transferase center (PTC), where the peptide bonds are formed, is located deep within the ribosome in its large (50S) subunit and is formed by RNA only. The newly synthesized peptide, also called the nascent chain (NC), exits the ribosome through a tunnel spanning the 50S subunit (Fig. 1). The tunnel is about 10 nm long. Already the first structural studies suggested that the exit tunnel may facilitate secondary structure formation of the NC. It was suggested that the NC may fold or pre-fold within the exit tunnel. Only in recent years has evidence for this co-translational protein folding emerged [2]. Despite this progress many questions remain unanswered.
The exit tunnel is mostly formed by ribosomal RNA which carries a highly negatively charged backbone. In contrast, the RNA nucleobases are rather hydrophobic. Two proteins contribute to the tunnel wall in its innermost part, namely proteins L4 and L22. These form a constriction within the tunnel and have been suggested to play some regulatory role. Some peptide sequences cause ribosome stalling; these stalled peptides can be released under certain conditions. Stalling is also of key importance for the action of many antibiotics. It means that the tunnel is a functional part of the ribosome. Moreover, the tunnel is filled with water and ions. The interaction between the NC and the complex heterogeneous environment of the interior of the tunnel are not currently understood.
In the project we used all-atom molecular dynamics (MD) simulations to study NC-ribosome complexes. We have developed a simulation protocol to study NC elongations. One by one, amino acids were added to the NC and its passage through the tunnel was investigated. Apart from several model peptide sequences we have studied a NC called VemP which was recently shown to regulate ribosome function.
VemP is a short peptide found in marine bacteria. Its structure has recently been resolved by a cryo-electron microscopy at atomic resolution. In the ribosome tunnel VemP may adopt an extremely compact conformation of two α-helices joined by an S-shape loop. By means of all-atom MD simulations we have studied the dynamics and mechanical properties of VemP.
Our simulations pose several challenges which can be tackled only by high-performance supercomputer facilities. To address biologically relevant questions about ribosome structure and dynamics, the simulations need to have high spatial resolution and the simulation time needs to be sufficiently long [3]. Hence our typical simulations contain more than 2 million particles whose time evolution is studied on multi-microsecond time scales.
Results and Methods:
Model Peptides
We studied three model peptide sequences: poly-phenylalanine (poly-Phe), poly-alanine (poly-Ala), and poly-glycine (poly-Gly), which we have built on-the-fly using the MD-based simulation protocol and various simulation setups. Motivated by experiments performed by our in-house collaborators (Marina Rodnina group), some of the peptides were labeled by a fluorescent dye BOF. We observed slow relaxation of the poly-Phe, so only a chain of 6 amino acids was technically possible to accommodate into the tunnel during the simulations. Poly-Gly and poly-Ala simulations relaxed faster than poly-Phe, possibly due to their smaller side chains. We were able to build from scratch a NC containing 16 amino acids. The pushing force, generated during the first steps of the amino acid addition in the simulations, dissipated quickly and did not propagate to distances beyond a few amino acids. As a consequence the NC formed a compact fold already before the constriction site (Fig. 2). Overall, our simulations suggest that the pushing force, exerted to the C-terminus of the NC, cannot be solely responsible for the peptide passage through the tunnel.

Figure 2: The compact structure of the BOF-labeled poly-alanine (yellow) in the ribosome tunnel (white surface). The P-site tRNA is in red.
Copyright: MPI BPC, GöttingenVemP
We used MD simulations to study VemP dynamics. The initial atomic structure was kindly provided by the group of Roland Beckmann (LMU Munich). All of the VemP components were stable at the simulations time scales (up to microseconds). The outer helix showed higher flexibility than the inner helix (Fig. 3). In experiments VemP reacts to external mechanical forces. Hence we have carried out a series of non-equilibrium simulations to study mechanical properties of the VemP. We applied a force onto the VemP N-terminus directing towards the tunnel exit. We tested several parameters that define the unfolding rate. Our simulations suggested that the unfolding triggered by the external mechanical force on the N-terminus occurs in a stepwise manner. VemP, as a mechanical string, is loaded until intermolecular contacts are broken suddenly, which causes unfolding of a VemP part (Fig. 3).
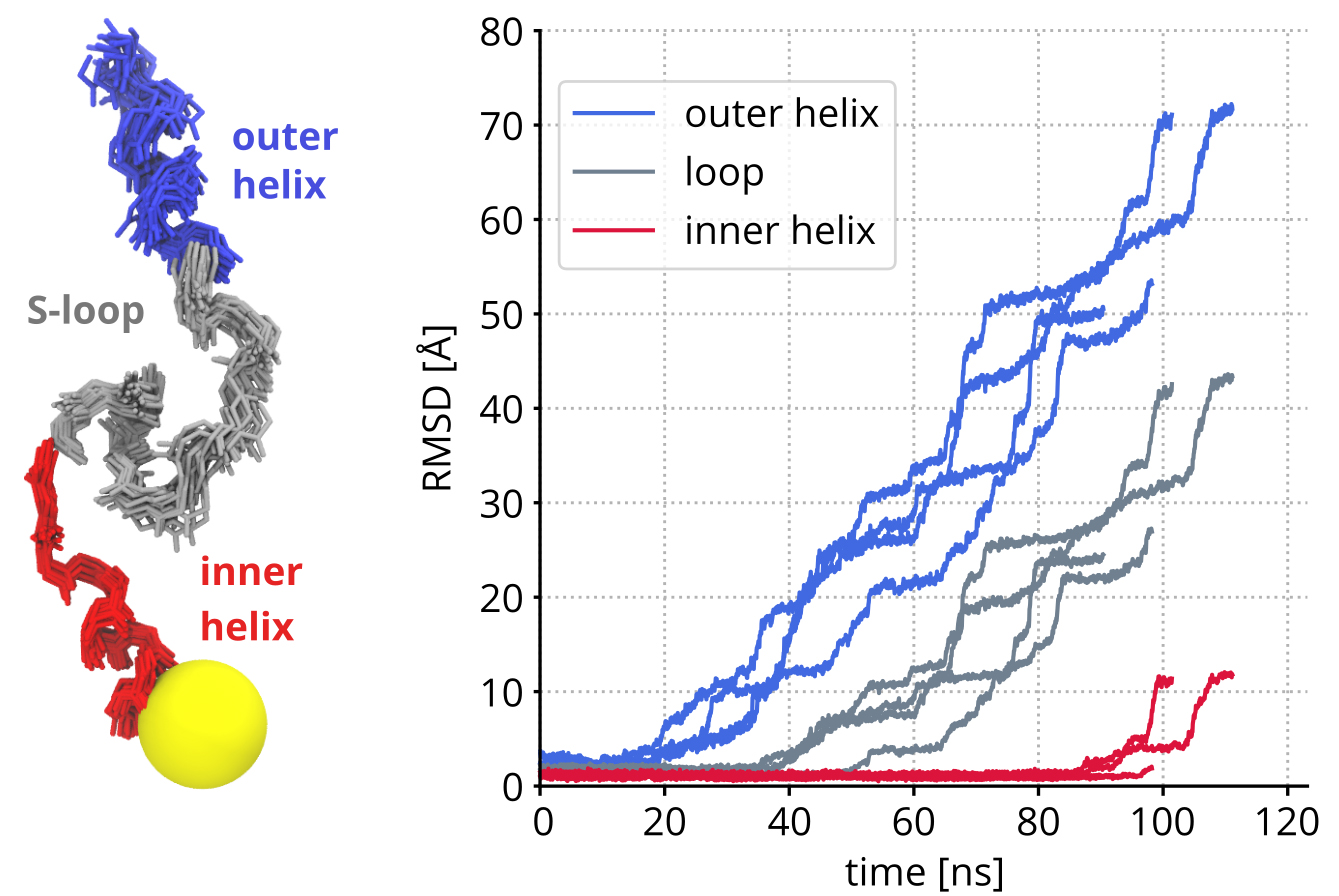
Figure 3: Left: Superimposition of 60 VemP conformations from the last 200 ns of the ribosome-VemP complex simulation. The PTC is represented by the yellow sphere Right: Root-mean-square deviation (RMSD) of VemP parts calculated as a function of simulation time with respect to the initial (i.e. experimental) VemP conformation. Four curves of the same color stand for four independent non-equilibrium simulations.
Copyright: MPI BPC, GöttingenMethods
All of the simulations were performed in GROMACS 5 package [4], which is available as a standard SuperMUC module. The code uses a mixed MPI/OpenMP parallelization and with our simulation boxes scales well up to several thousands of CPU cores. The simulations ran in chains using a checkpoint file based functionality of GROMACS. The simulations produced a large amount of data (tens of TB) which are still being analyzed.
On-going Research / Outlook
We have studied several NCs in the ribosome exit tunnel. Many aspects of the peptide elongation remain to be addressed. In order to avoid formation of a compact NC fold, we will combine two approaches that were used in the current Project, namely the pushing force generated by addition of an extra amino acid to the C-terminus with the pulling force applied to the N-terminus of the NC. Regarding the VemP peptide, we plan to study its mechanical properties in more detail. Molecular details of the unfolding will likely emerge from the analyses of the now-available trajectories.
Research Team
Michal H. Kolář, Lars. V. Bock, Andrea C. Vaiana
Project Partners
Dept. Phys. Biochemistry, Max Planck Institute for Biophysical Chemistry (MPI BPC), Göttingen; Gene Center, Ludwig-Maximilians-Universität (LMU), Munich
References and Links
[1] Jacob Poehlsgaard and Stephen Douthwaite. 2005. The bacterial ribosome as a target for antibiotics. Nature Reviews Microbiology, 3 (Nov 2005), 870-881.
[2] Boyd Hardesty, Tamara Tsalkova and Gisela Kramer. 1999. Co-translational folding. Curr. Opin. Struct. Biol., 9 (Feb 1999), 111-114.
[3] Lars V. Bock, Michal H. Kolář, and Helmut Grubmüller. 2018. Molecular simulations of the ribosome and associated translation factors. Curr. Opin. Struct. Biol., 49 (April 2018), 27-35.
[4] Mark J. Abraham, Teemu Murtola, Roland Schultz, Szilard Pall, Jeremy C. Smith, Berk Hess and Erik Lindahl. 2015. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software X. 1 (Sep. 2015), 19-25.
Scientific Contact:
Prof. Dr. Helmut Grubmüller
Theoretical and Computational Biophysics Dept.
Max-Planck-Institute for Biophysical Chemistry
Am Fassberg 11, D-37077 Göttingen (Germany)
e-mail: hgrubmu[at]gwdg.de
NOTE: This report was first published in the book "High Performance Computing in Science and Engineering – Garching/Munich 2018".
LRZ project ID: pr62de
April 2019