
MATERIALS SCIENCE AND CHEMISTRY
High Accuracy Molecular Dynamics Simulation of Fluids at Interfaces
Principal Investigator:
Martin Horsch
Affiliation:
Laboratory of Engineering Thermodynamics, University of Kaiserslautern (Germany)
Local Project ID:
pr83ri
HPC Platform used:
SuperMUC of LRZ
Date published:
At molecular resolution, the interface which separates coexisting fluid phases, e.g. between a liquid droplet and a surrounding vapour, is not sharp but continuous. Both the interface and the coexisting phases are subject to significant fluctuations which also contribute to the surface tension. Therefore, it is crucial to accurately reproduce the surface tension and analyse the properties of nanodroplets with a method that resolves the structure of fluid matter at the molecular level. Here, molecular dynamics (MD) simulation is applied to analyse interfacial phenomena qualitatively and capture interfacial properties quantitatively.
For MD codes, as for any software, there is a trade-off between generality and optimality for a single purpose, which no particular implementation can completely evade. The program ls1 mardyn (large systems 1: molecular dynamics) expands the temporal and spatial range of scales accessible to molecular simulation, with a focus on inhomogeneous systems (e.g. at interfaces) and non-equilibrium thermodynamics [1]. It is available as free software [2] and was applied and extended within the present project to accurately and efficiently simulate systems with vapour-liquid interfaces.
By massively-parallel high performance computing, e.g. using the ls1 mardyn program, molecular simulation becomes an experiment in silico. Due to their hetero-geneous structure, systems with a phase boundary have stronger long-range interactions, which also need to be taken into account [3]. Load balancing by recursive bisection of the simulation volume, using a linked-cell data structure, significantly improves the scalability of the code. In this way, supercomputers with a large number of cores can be used efficiently both for homogeneous and heterogeneous systems [1].
Results and Methods
Molecular simulations with system dimensions that far exceed the cut-off radius, beyond which a mean-field approach is employed for the intermolecular interactions, are most efficiently parallelized by space decomposition schemes. Thereby, the simulation volume is subdivided into smaller subvolumes (one for each process) that ideally carry the same load. In ls1 mardyn, an interface class for the domain decomposition scheme permits the generic implementation of different load balancing strategies operating on spatial subdomains with a linked-cell data structure [1].
To make use of hyperthreading, a lightweight shared-memory parallelization was implemented: By employing a sliding window, as sketched in Fig. 1, including prefetched cells, two threads can operate concurrently on independent cells. Thereby, it is avoided that threads work on directly neighbouring cells simultaneously. Therefore, a barrier, causing comparably little overhead on a hyperthreading core, is required after each thread has processed a cell. This allows the execution of one MPI rank per core with two OpenMP threads to create sufficient instruction level parallelism, leading to a significant performance improvement on SuperMUC.
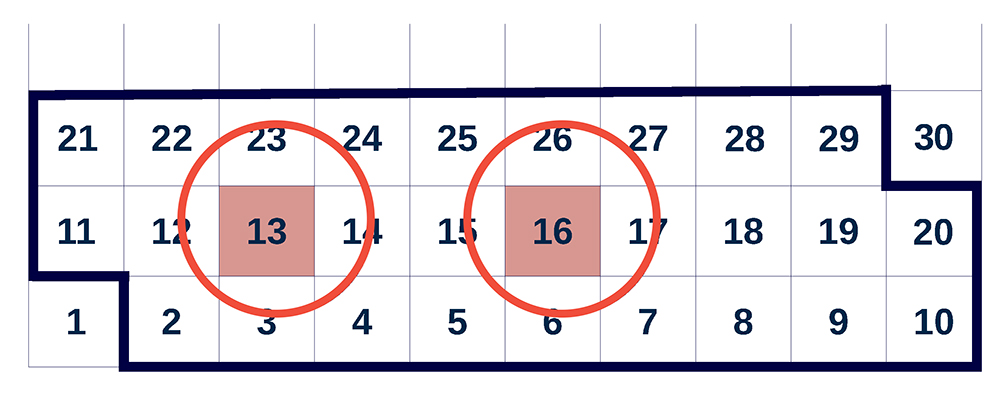
Figure 1: Hyperthreaded sliding window (represented here in two dimensions, instead of three as in the actual code), employed by the ls1 mardyn version optimized for the Intel Sandy Bridge EP architecture. At the present stage, the two highlighted cells 13 and 16 are concurrently processed by SMT threads. Thereby, cell 2 is considered as a neighbour cell for the last time, leaving the sliding window, and cell 27 enters the sliding window; cells 28 and 29 are prefetched.
Copyright: LTD, University of KaiserslauternMolecule data outside the sliding window are stored in the form corresponding to the object-oriented software architecture, i.e. as a dynamic array of structures (AoS) containing Molecule objects. When the sliding window is shifted further and covers a new cell, the positions and velocities of the molecules in that cell are converted to a structure of arrays (SoA) form suitable for vectorization.
The two-centre Lennard-Jones plus point quadrupole (2CLJQ) class of molecular models provides a straightforward description of the intermolecular interactions for many low-molecular compounds [3]. Furthermore, vapour-liquid interfaces – for systems with large numbers of molecules – can for this model class be efficiently treated using a long-range correction that was developed within the present project, yielding a high accuracy for the computed surface tension. Accordingly, simulations of the vapour and liquid phases in direct coexistence were carried out along the whole vapor pressure curve, and the model parameters were varied systematically, covering the whole parameter range of 2CLJQ models for real fluids available from the literature.
Based on the simulation results for the surface tension, a precise correlation was obtained in dependence of the parameters, which is universally valid for the whole model class. In combination with the available knowledge of bulk fluid properties at vapour-liquid equilibrium conditions, this correlation can be used to adjust 2CLJQ models to multiple types of real fluid properties, e.g. by multi-criteria optimization based on an evaluation of the Pareto set, cf. Fig. 2, where this is illustrated for carbon dioxide using three criteria: Average agreement for the saturated liquid density, the vapour pressure, and the surface tension [3].
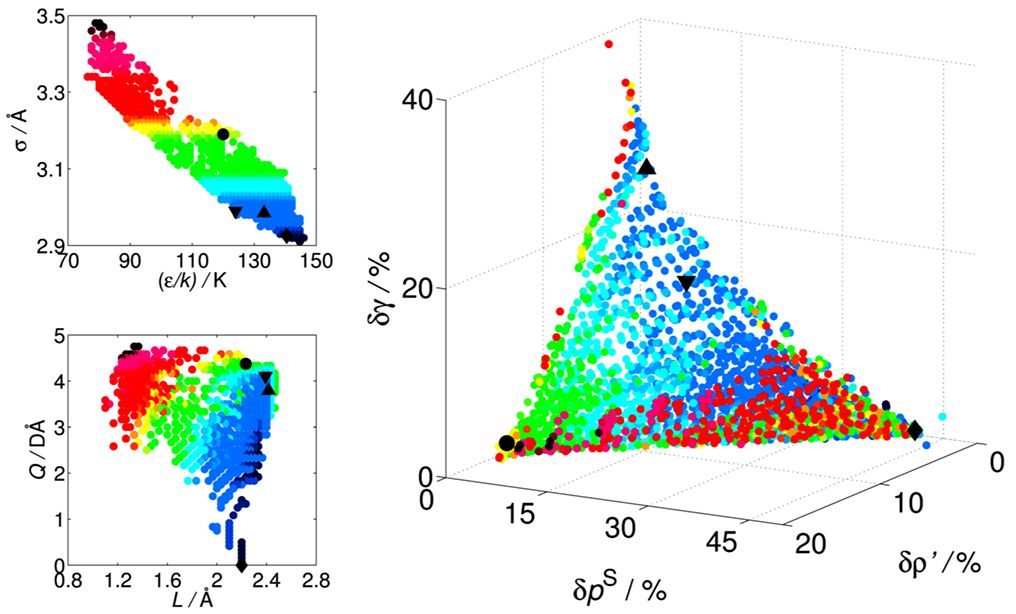
Figure 2: Pareto set within the parameter space (left) of the 2CLJQ class for modelling carbon dioxide as well as its representation in the objective space (right), taking three criteria into account, i.e. the average deviation of (correlated) model properties from (correlated) experimental data for the saturated liquid density, the saturated vapour pressure, and the vapour-liquid surface tension [3].
Copyright: LTD, University of KaiserslauternFurthermore, three-phase contact between the vapour-liquid interface of a sessile droplet and a solid surface was systematically investigated for the truncated-shifted Lennard-Jones potential (LJTS), considering the scenario with a perfectly planar solid substrate, cf. Fig. 3, as a reference case for future work on patterned and rough surfaces. Both the fluid and the solid were modelled by the LJTS potential (with different parameters), varying both the solid density and the dispersive fluid-solid interaction energy. From the present simulation results, a universal correlation was derived for the contact angle as a function of the well depth of the unlike fluid-solid interaction and further characteristic parameters, which was found to carry over to other similar systems with a good accuracy [4].

Figure 3: Snapshots from simulations of sessile droplets at the same temperature on walls of the same density and structure, differing only in the magnitude of the dispersive fluid-solid interaction, which is larger on the right side, yielding increased wetting, i.e. a smaller contact angle [4].
Copyright: LTD, University of KaiserslauternOn-going Research / Outlook
The present project partners LTD (Kaiserslautern), SCCS (Munich), and ThEt (Paderborn) have applied for a follow-up computing project entitled Scalable, Performant and Resilient Large-scale Applications of Molecular Process Engineering (SPARLAMPE), which is expected to start in the first quarter of 2016.
Based on the approaches for high-accuracy simulations established within the present computing project, a more direct orientation towards applications in mechanical process engineering has become feasible, based on the scalable and performant implementation of algorithms for heterogeneous sytems in ls1 mardyn. For validated and reliable molecular models, the SPARLAMPE project will further focus on linear transport coefficients in the bulk fluid and at interfaces as well as transport processes near and far from equilibrium.
These are necessarily HPC applications, since finite-size effects need to be assessed rigorously and the computation of transport properties requires a much more extensive sampling than other thermodynamic properties. In particular, as larger systems also require a longer relaxation time to reach equilibrium, fast equilibration techniques will be developed, implemented and employed for petascale simulations.
Research Team:
Martin Horsch1, Stefan Becker1, Katrin Stöbener1, Stephan Werth1, Stefan Eckelsbach2, Wolfgang Eckhardt3, Alexander Heinecke3, Nikola Tchipev3, Hans-Joachim Bungartz3, Jadran Vrabec2, Hans Hasse1
1Laboratory of Engineering Thermodynamics, University of Kaiserslautern
2Thermodynamics and Energy Technology, University of Paderborn
3Scientific Computing in Computer Science, Technische Universität München
References and Links
[1] Niethammer, C., Becker, S., Bernreuther, M., Buchholz, M., Eckhardt, W., Heinecke, A., Werth, S., Bungartz, H.-J., Glass, C. W., Hasse, H., Vrabec, J., and Horsch, M., J. Chem. Theory Comput. 10(10): 4455-4464 (2014).
[2] http://www.ls1-mardyn.de/
[3] Werth, S., Stöbener, K., Klein, P., Küfer, K.-H., Horsch, M., Hasse, H., Chem. Eng. Sci. 121: 110-117 (2015).
[4] Becker, S., Urbassek, H. M., Horsch, M., and Hasse, H., Langmuir 30(45): 13606-13614 (2014).
Scientific Contact:
Jun. Prof. Dr.-Ing. Martin Horsch
Laboratory of Engineering Thermodynamics, University of Kaiserslautern
Erwin-Schrödinger-Str. 44, D-67663 Kaiserslautern (Germany)
e-mail: martin.horsch [@] mv.uni-kl.de
February 2016