
MATERIALS SCIENCE AND CHEMISTRY
Chemical Functionalization of Oxide Surfaces
Principal Investigator:
Bernd Meyer
Affiliation:
Interdisciplinary Center for Molecular Materials and Computer-Chemistry-Center, Friedrich-Alexander-Universität Erlangen-Nürnberg (Germany)
Local Project ID:
pr74be
HPC Platform used:
SuperMUC of LRZ
Date published:
The electronic and optical properties of oxide surfaces and nanoparticles can be tuned by attaching specifically tailored organic molecules. This is employed in molecular electronics or when building dye-sensitized solar cells. Such a chemical functionalization is usually done in solution. In this work, advanced molecular dynamics sampling techniques based on a quantum-chemical description of the atomic interactions are used to obtain a fundamental understanding of the chemical reaction mechanisms at such solid-liquid interfaces. The simulations allow to identify the key reaction intermediates and they provide new insights into the important role of the hydrogen-bond network and the mobility of protons at the interface.
Introduction
The functionalization of oxide surfaces by chemical attachment of molecules plays an important role in nanoparticle synthesis, molecular electronics, fabrication of hybrid organic/inorganic solar cells and many other areas of technological interest. A shell of molecules can protect the underlying surface from chemical attack, it can be used to tune material's properties, e.g. the work function or electron injection barrier, or it can act as functional unit itself, e.g. as conducting channel in a molecular field-effect transistor or as dye in a organic/inorganic solar cell.
Surface functionalization is mostly done by wet-chemical processes. Molecules typically attach by condensation reactions, i.e. by elimination of water molecules. While chemical surface reactions with molecules from the gas phase have been studied extensively by quantum chemical calculations, mechanisms of chemical reactions at the solid/liquid interface are basically unexplored.
By using molecular dynamics (MD) simulations we aim at a fundamental understanding of the elementary reaction steps when molecules bind from solution to a surface. In particular, we address the role of the solvent and the impact of the surface structure and composition on the reaction mechanisms. For our simulations we have chosen alumina (Al2O3) as a prototypical oxide substrate, isopropanol as the liquid phase and methylsilanetriol (MST) as reactive molecule.
Results and Methods
For an unbiased description of the breaking and formation of chemical bonds in the chemical processes of surface functionalization we use ab initio molecular dynamics (AIMD) simulations, specifically the Car-Parrinello molecular dynamics (CPMD) method and code [2]. CPMD is based on density functional theory (DFT) as quantum-chemical electronic structure method. To overcome reaction barriers and to be able to observe chemical reactions (`rare events’) in the limited timeframe of AIMD simulations we apply various accelerated sampling techniques, in particular thermodynamic integration, umbrella sampling and metadynamics (MTD), which also provide information on the free energy landscape.
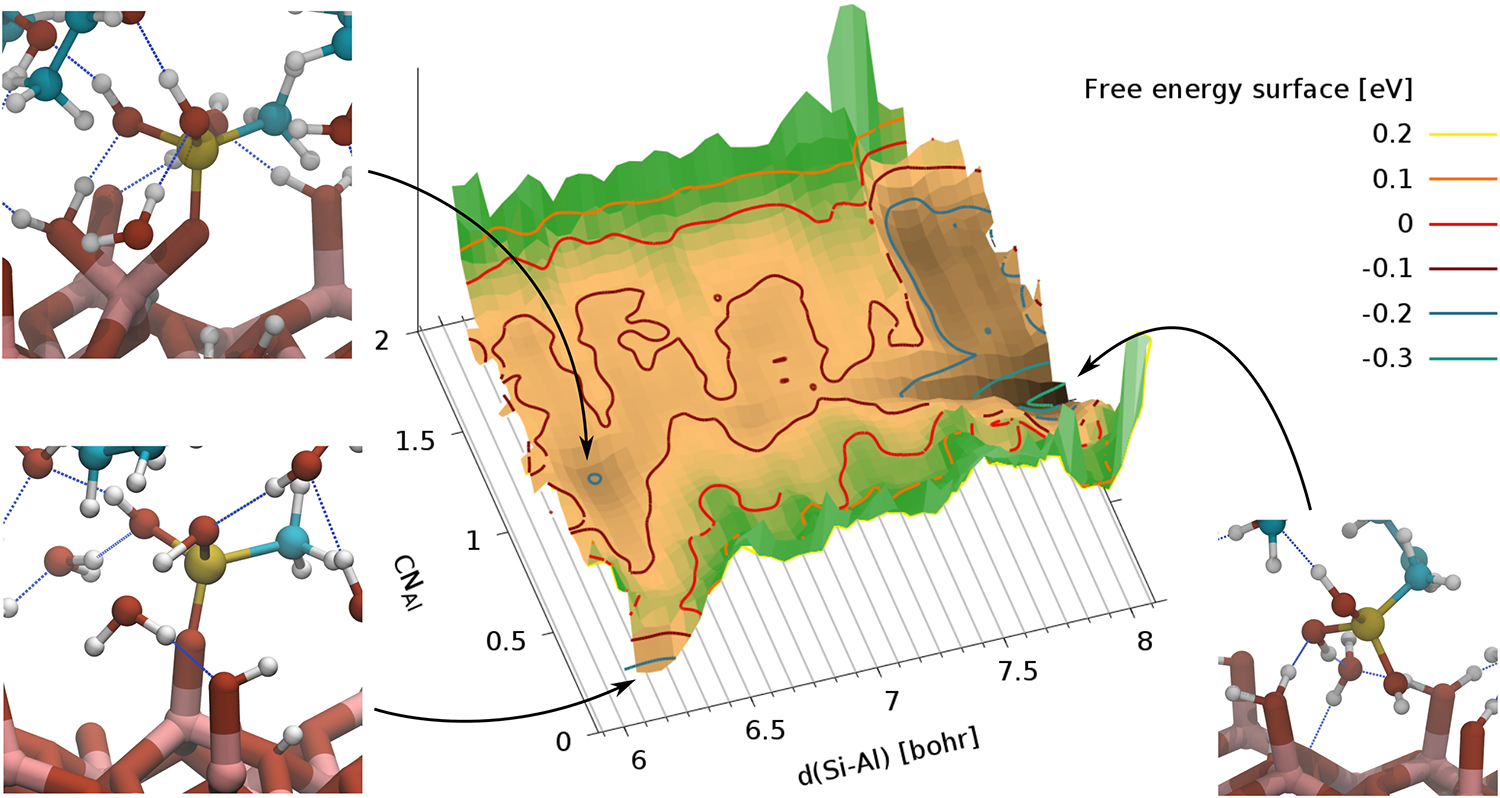
Figure 1: Free-energy landscape from a WS-MTD simulation. The intact MST molecule approaches the alumina surface from the isopropanol solution (right inset). One of the three OH groups of MST coordinates to a surface Al site with an adsorbed OH group and deprotonates (upper left inset). In the final step, the OH group at the Al site is protonated and desorbs as a water molecule from the Al site (lower left inset). The umbrella sampling was done for the Al Si distance. The positions of the umbrella potentials are indicated by gray lines. The coordination number (CN) of the Al adsorption site with all oxygen atoms (except those from MST) was used as collective variable in metadynamics. The unit cell contains 503 atoms and 1462 electrons. Al, O, C, Si and H are shown in light red, dark red, blue, yellow and white, respectively. Hydrogen bonds are indicated by blue dotted lines.
Copyright: ICMM/CCC, FAU Erlangen-Nürnberg (Germany)The simulations show that the approach of MST molecules from the liquid to the alumina surfaces is an activated process with a small energy barrier. One of the MST OH groups reorients from the liquid towards the surface and coordinates to a surface Al ion. A very characteristic transition state structure of a six-membered ring is formed, which consists of the Si atom and one OH of the MST molecule and the surface Al with an adsorbed OH group. The transition structure quickly leads to a spontaneous condensation reaction in which the OH at the Al site is protonated and desorbs as water molecule into the liquid.
Residual water molecules on the Al2O3 surface increase the energy barrier for surface binding. Also the condensation reaction becomes activated. An example of the overall free energy landscape for this process is shown in Figure 1. Here we have used the recently proposed well-sliced metadynamics (WS-MTD) technique [3], a combination of umbrella sampling and metadynamics, which allows us to study the full process in a single simulation setup.
On the multi-core architecture of SuperMUC with 28 cores per node the MPI-only parallelization of CPMD came to its limits. First, the reduction in band width and increase in overhead of the all-to-all and global summation calls of 28 MPI processes communicating simultaneously from each node became a severe bottleneck. Second, for our isopropanol/Al2O3 system scalability already ends at 7 nodes (with 2 ps simulation time per day), since then the fast Fourier grid is fully distributed over the MPI processes (see red dashed line in Figure 2). 50 ps of simulation time, which is a typical length of a production run, would therefore take at minimum about a month.
In a KONWHIR project in collaboration with Gerald Mathias from LRZ we optimized the parallelization strategies within CPMD. This included the following steps: (i) the existing OpenMP parallelization is extended to all relevant code paths, (ii) single node performance is further increased by merging various small DGEMM calls into larger ones which are called less frequently, (iii) the communication overhead is reduced by introducing overlapping computation and communication and by modifying the all-to-all message passing in such a way that larger fragments of data are communicated less frequently.
The result is shown in Figure 2 (solid red line). We typically use 4 MPI processes per node and 7 OpenMP threads per MPI process. Our example of the isopropanol/Al2O3 interface shows very good scaling up to 25 nodes with the new improved CPMD version. On 12 nodes we get more than 10 ps simulation time per day, i.e. a 50 ps production run can be done now within 5 days instead of one month. On the single node level, when staying in the range of 2 to 6 nodes where CPMD shows almost ideal scaling, we achieve about 50% of the node peak performance.

Figure 2: Improved performance of CPMD on SuperMUC. The small system is the isopropanol/Al2O3 simulation from Figure 1, the large system is the water/ZnO interface from Figure 3.
Copyright: ICMM/CCC, FAU Erlangen-Nürnberg (Germany)On-going Research / Outlook
With the improved scalability and single node performance of CPMD we are now able to address more complex chemical processes at oxide surfaces. In a follow-up project we have started to study the catalytic deactivation of chemical warfare agents at ZnO surfaces in aqueous environments [4]. A typical setup is shown in Figure 3. By using 36 SuperMUC nodes we are able to perform 50 ps simulations for this system within 10 days (about 250,000 CPUh, see blue solid line in Figure 2). In the largest simulation so far we carried out an umbrella sampling run to calculate the adsorption free energy of sulfur mustard molecules on ZnO. For each umbrella window we used 18 nodes. The 28 umbrella windows were run in parallel to enhance sampling by replica exchange, thus making use of a full SuperMUC island with 512 nodes.

Figure 3: Sulfur mustard molecule at a water/ZnO interface. The unit cell contains 735 atoms and 3332 electrons. Zn, O, C, S, Cl and H are shown in gray, red, black, yellow, green and cyan, respectively.
Copyright: ICMM/CCC, FAU Erlangen-Nürnberg (Germany)Research Team
Tobias Klöffel, Bernd Meyer (PI), Paul Schwarz
Project partners
KONWHIR, Leibniz Supercomputing Centre (LRZ), Garching/München

Figure 04: Snapshot from an ab inito molecular dynamics simulation of the adsorption of a methylsilanetriol molecule from isopropanol solution to an alumina surface.
Copyright: ICMM/CCC, FAU Erlangen-Nürnberg (Germany)References and Links
[1] https://chemistry.nat.fau.eu/meyer-group
[2] http://www.cpmd.org
[3] S. Awasthi, V. Kapil, N.N. Nair, J. Comp. Chem. 99, 12562 (2016)
[4] G.K. Prasad et al., J. Hazard. Mater. 149, 460 (2007)
Scientific Contact:
Prof. Dr. Bernd Meyer
Interdisciplinary Center for Molecular Materials (ICMM)
and Computer-Chemie-Centrum (CCC)
Friedrich-Alexander-Universität Erlangen-Nürnberg
Nägelsbachstraße 25, D-91052 Erlangen (Germany)
e-mail: bernd.meyer [@] chemie.uni-erlangen.de
NOTE: This report was first published in the book "High Performance Computing in Science and Engineering – Garching/Munich 2018".
LRZ Project ID: pr74be
April 2019