
MATERIALS SCIENCE AND CHEMISTRY
First-Principles Modeling of Minerals, Melts and Fluids at High Pressures and High Temperatures
Principal Investigator:
Sandro Jahn
Affiliation:
Institute of Geology and Mineralogy, University of Cologne (Germany)
Local Project ID:
hpo15
HPC Platform used:
JUQUEEN of JSC
Date published:
Comprehension of processes in the deep Earth’s interior requires knowledge of the structure and properties of geologically relevant materials at high pressures and high temperatures. In this project, first-principles molecular dynamics simulations are employed to complement experimental efforts to study mainly structurally disordered materials under extreme conditions. For instance, in a recent study the structure of SiO2 glass was studied up to pressures of more than 1.5 Mbar. Further, the feasibility of predicting element partitioning between melts from first-principles has been explored.
Minerals, metal and oxide melts, and aqueous fluids are the principal constituents of the Earth. Understanding their atomic structure, physical and thermodynamic properties and chemical interactions at the high pressure and high temperature conditions of the Earth’s interior poses a major challenge in geomaterials research. As the deep Earth is largely inaccessible to direct sampling, our current knowledge about the structure and dynamics of our planet is mainly obtained from combining geophysical measurements with laboratory experiments and numerical simulations. Melts and fluids are especially active agents in geological processes but the investigation of their structure and properties at extreme conditions is very difficult.
In this project, the researchers use first-principles molecular dynamics approaches to predict structure, physical and thermodynamic properties of model systems of materials based on a quantum-mechanical treatment of atomic interactions. Such simulations are computationally extremely demanding and require the usage of high-performance computing (HPC) resources. Typically, simulation cells contain between one and a few hundred atoms and the molecular dynamics is sampled for tens to hundreds of picoseconds, which is a few orders of magnitude longer than the time needed for a single molecular vibration. The simulations are performed using the CPMD code that is optimized for the use on big supercomputers with a hybrid MPI-OMP parallelization.
SiO2 is a prototype model system for silicates. At room pressure the structure of silicate minerals, melts and glasses is characterized by tetrahedral SiO4 building blocks. From high-pressure phase transitions of crystalline silicates it is known that the coordination of Si changes from tetrahedral to octahedral between 10 and 25 GPa. In melts and glasses a continuous transition from 4-fold to 6-fold is observed in the pressure range from 15 to 40 GPa. The evolution of silicate melt structures at pressures above 100 GPa has remained elusive for a long time. Such extreme conditions are not only relevant for the Earth’s interior (e.g. 136 GPa at the core-mantle boundary) but also for other planetary bodies.
Very recently, the research team succeeded to perform the first successful x-ray diffraction study to reveal the structure of SiO2 glass up to pressures of 172 GPa (Prescher et al., 2017). The experiments and complementary first-principles molecular dynamics simulations showed that at pressures above 50 GPa the average coordination number of Si exceeds six to reach 6.8 at the highest experimental pressure and 7.3 at about 350 GPa in the simulations (see Figure). It is interesting to note that the Si-O nearest neighbor distance initially increases with increasing pressure up to about 30 to 40 GPa, which indicates that compression is mainly driven by increasing the coordination number. At higher pressures compression is also accompanied by shortening of Si-O bonds. Structural changes beyond 6-fold coordinated Si have been rationalized using the concept of oxygen packing fraction (Prescher et al., 2017).
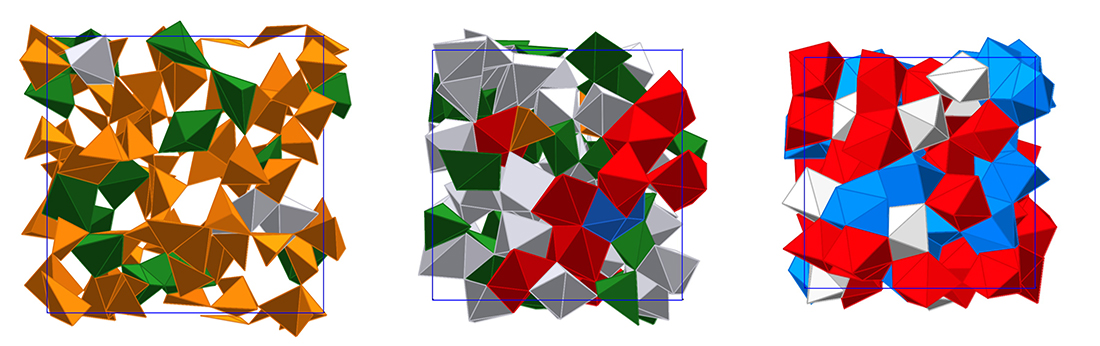
Snapshots from first-principles molecular dynamics simulations of SiO2 melt/glass at a temperature of 4000 K and densities of 2.66, 4.40 and 7.0 g/cm3. Si coordination environments are colored: 4 – orange, 5 – green, 6 – grey, 7 – red, 8 – blue.
Copyright: IGM, University of CologneAnother recent research activity is aimed at predicting element partitioning between different phases from first-principles simulations. Chemical elements are redistributed in the course of many geological processes. Measurements of (trace) element concentrations in rocks and fluid phases are frequently used to reconstruct timing and thermodynamic conditions of these processes. The biggest open question is how melt or fluid structure and composition determines partitioning behavior during mineral-melt or mineral-fluid interactions. In the simulations, the researchers use the method of thermodynamic integration to predict small differences in Gibbs energies during exchange reactions. Derived estimations of partition coefficients indicate the high potential of this approach (Künzel et al. 2017; Wagner et al. 2017).
This research is a first step towards the development of new models for element partitioning, which are strongly demanded by the geochemistry community.
References:
Künzel, D., Wagner, J., Jahn, S., 2017. Ni partitioning between metal and silicate melts: an exploratory ab initio molecular dynamics study, Chem. Geol. 461, 47-53.
Prescher, C., Prakapenka, V.B., Stefanski, J., Jahn, S., Skinner, L.B., Wang, Y., 2017. Beyond sixfold coordinated Si in SiO2 glass at ultrahigh pressures. PNAS 114, 10041-10046.
Wagner, J., Künzel, D., Haigis, V., Jahn, S., 2017. Trace element partitioning between silicate melts - a molecular dynamics approach, Geochim. Cosmochim. Acta 205, 245-255
Scientific Team:
Prof. Dr. Sandro Jahn (PI), Dr. Clemens Prescher, Johannes Stefanski
Scientific Contact:
Prof. Sandro Jahn
Institute of Geology and Mineralogy (IGM)
University of Cologne
Zülpicher Str. 49b, D-50674 Köln (Germany)
e-mail: s.jahn[at]uni-koeln.de
November 2017