
MATERIALS SCIENCE AND CHEMISTRY
Quantum-Powered Drug Discovery: Advancing Molecular Simulations with High- Performance Computing
Principal Investigator:
Dr. Emiliano Ippoliti
Affiliation:
Forschungszentrum Jülich GmbH, INM-9 Institut für Neurowissenschaften und Medizin - Computational Biomedicine, Jülich, Germany
Local Project ID:
idh1
HPC Platform used:
JUWELS CPU at JSC
Date published:
Abstract
This project uses quantum-powered computer simulations to improve how new drugs are discovered. Traditional simulation methods often fail for complex proteins, especially those with metals or chemical reactions. By combining quantum and classical physics in the newly developed MiMiC framework and running on the JUWELS supercomputer at Jülich, a new Quantum HPC Virtual Screening (QHPC–VS) method was developed. Applied to the mutant IDH1 enzyme linked to brain cancer, it identified 15 potential PET imaging tracers, offering new tools for faster, non-invasive diagnosis.
Report
Discovering a new drug is one of the most complex challenges in modern science. The process, from the first idea to an approved medicine, can take over a decade and cost billions of euros. One of the most time-consuming stages is the preclinical phase: early research that identifies and refines potential drug molecules before they ever reach clinical testing. To shorten this process, scientists increasingly rely on computer simulations to predict how drug candidates will interact with their target proteins inside the human body. These computational tools allow researchers to test thousands of compounds in silico, i.e., virtually, before conducting expensive and time-consuming experiments in the lab.
One of the most successful techniques in this field is called structure-based drug design, which uses detailed three-dimensional structures of biological targets to design molecules that can bind to them. Another key approach is molecular dynamics (MD), a computer method that simulates how atoms of these targets move over time according to the laws of physics. Together, these techniques allow scientists to visualize and predict how potential drugs might fit into and affect their target proteins. This then allows for the scientific design of drugs.
However, despite these powerful tools, current simulation methods still have significant limitations, particularly when dealing with complex biological systems such as metal-containing enzymes or transition states (temporary molecular arrangements during chemical reactions). These are precisely the kinds of systems that are crucial in many therapeutic areas, and where more accurate methods are urgently needed.
Traditional molecular dynamics simulations rely on classical physics, where atoms are treated as solid spheres connected by spring-like bonds. The forces that determine their motion are derived from mathematical equations known as force fields. These force fields are carefully parameterized to reproduce experimental data for simple molecules and proteins.
While classical MD has become a routine tool in drug discovery, it faces fundamental challenges when trying to describe chemical processes that involve bond breaking, bond formation, or electron transfer. Such phenomena are inherently quantum mechanical in nature; they depend on the behavior of electrons, which cannot be captured by classical physics alone. Moreover, proteins that contain metal ions (such as iron, zinc, or magnesium) play essential roles in catalysis and regulation, but metals have complex electronic structures that classical force fields cannot accurately model. As a result, classical MD simulations can produce unrealistic geometries or even distort the active sites of these proteins, precisely where potential drugs are supposed to bind. This limitation has restricted the use of molecular dynamics in certain key areas of pharmaceutical research. To move beyond these barriers, researchers are turning to more sophisticated methods that explicitly consider quantum mechanical effects.
Quantum mechanics (QM) describes the behavior of electrons and atoms at the smallest scales. By solving the equations of quantum physics—most commonly through an approach called Density Functional Theory (DFT) —scientists can calculate molecular energies and electronic properties with high accuracy. Unfortunately, these calculations are computationally demanding. A fully quantum description of an entire protein (which might have tens of thousands of atoms) would require an unrealistic amount of time and computer power, even on the world’s fastest supercomputers. To address this, scientists have developed hybrid methods that combine quantum mechanics with classical molecular mechanics (MM). These are called QM/MM methods. In this approach, only the most critical part of a biological system—such as the enzyme’s active site where reactions take place and the drug binds—is treated using quantum mechanics, while the rest of the protein and surrounding solvent are simulated using classical physics. This hybrid QM/MM approach allows researchers to study reactions and interactions in biological systems with much higher accuracy than classical MD alone, while keeping the computational cost manageable.
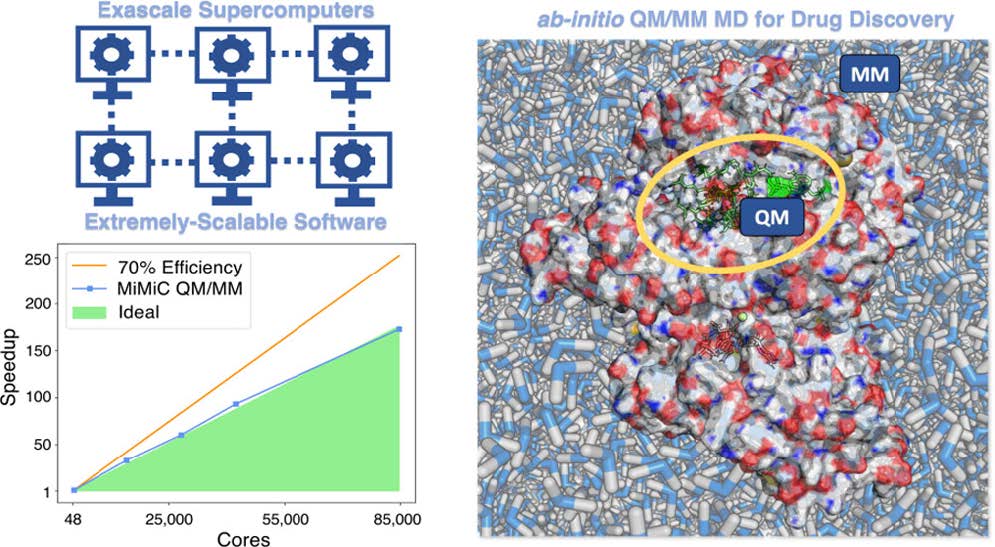
In recent years, a new computational framework called MiMiC (Multiscale Modeling in Computational Chemistry) was introduced to enable efficient and scalable QM/MM molecular dynamics simulations. MiMiC allows different specialized simulation programs to work together; for instance, coupling a quantum mechanical code like CPMD or CP2K with a classical molecular dynamics engine such as GROMACS. This design makes it possible to perform multiscale simulations where each region of the system is described at the appropriate level of theory. The MiMiC framework is specifically designed to take advantage of High-Performance Computing (HPC) systems, distributing the calculations across thousands of processors to achieve both speed and accuracy.
In this work, the authors explored how this MiMiC-based QM/MM molecular dynamics approach could be used to improve the realism and predictive power of drug discovery simulations, especially for biologically complex targets that classical methods struggle to describe. All simulations in this project were performed on JUWELS, the flagship supercomputer of the Jülich Supercomputing Centre (JSC) in Germany. JUWELS is one of the most powerful HPC systems in Europe, designed to handle large-scale scientific simulations requiring massive parallel computation. By leveraging JUWELS, quantum-level molecular dynamics simulations on large biomolecules were able to be carried out—something that would be impossible on conventional computers. The combination of MiMiC and JUWELS provided exceptional performance and scalability, demonstrating that QM/MM simulations can now be extended to real biological systems at a meaningful scale.
To make quantum mechanical simulations practical for drug discovery, a new computational workflow called the Quantum High-Performance Computing Virtual Screening (QHPC–VS) protocol was designed by the authors. Virtual screening is a process where thousands (or even millions) of molecules are computationally tested for their ability to bind to a target protein. Typically, this is done using fast but approximate methods like molecular docking and classical MD. The QHPC–VS pipeline extends this idea by incorporating quantum mechanics directly into the screening process.
To demonstrate the potential of this new pipeline, it was applied to a medically important protein: Isocitrate Dehydrogenase 1 (IDH1). IDH1 is an enzyme that plays a key role in cellular metabolism—specifically in the conversion of isocitrate to α-ketoglutarate. Mutations in the gene that contains the instructions for the cell to express the IDH1 enzyme are known to alter this normal function, leading to the production of an abnormal metabolite called 2-hydroxyglutarate in place of α-ketoglutarate. This compound accumulates in cells and interferes with gene regulation, contributing to the development of several types of cancer, including gliomas (a form of brain tumor) and acute myeloid leukemia.
A particular mutation, known as R132H, has become an important biomarker—a molecular signal that helps identify certain forms of glioma. Because this mutant form of IDH1 is found specifically in tumor cells but not in healthy tissue, it represents an ideal target for non-invasive imaging methods such as positron emission tomography (PET). PET imaging works by injecting the patient with a small molecule that contains a radioactive isotope, known as a radiotracer. This tracer binds selectively to a target in the body, and its radioactive signal is detected by the PET scanner to create an image of where the target is located. Developing a PET tracer for mutant IDH1 could enable doctors to visualize gliomas in real time and even track how they respond to therapy. However, designing such a tracer has proven extremely difficult. The main problem lies in the complex active site of the IDH1 enzyme. This region contains metal ions and undergoes subtle changes during catalysis; both of which are poorly represented by classical force fields. As a result, when scientists tried to model this site using traditional molecular dynamics, the structure became distorted and produced unreliable results. This is precisely the kind of challenge where quantum mechanical simulations can make a difference. By accurately describing the electronic structure of the enzyme’s active site, a QM/MM simulation can reveal how potential tracer molecules might interact with the mutant IDH1 enzyme at an atomic level.
Using the MiMiC framework and the QHPC–VS protocol, the authors performed detailed QM/MM molecular dynamics simulations of the R132H mutant IDH1 enzyme. These simulations modeled the enzyme’s active site quantum mechanically while treating the rest of the protein and solvent classically. Running these calculations on the JUWELS supercomputer allowed the system to achieve exceptional performance and scalability. The results demonstrated that MiMiC could accurately capture the structural and dynamic behavior of the IDH1 active site — something that classical MD could not. For the first time, it became possible to simulate the mutant enzyme’s environment in realistic detail, enabling a reliable search for molecules that might selectively bind to it.
With the quantum-based simulation pipeline established, a virtual screening campaign was conducted to identify small molecules that could act as selective ligands for the mutant IDH1 enzyme. These molecules were chosen based on their ability to bind to the active site in stable configurations during the QM/MM simulations. From this process, 15 promising candidates emerged as potential PET radiotracer precursors. Remarkably, among them was fluorothymidine—a molecule already well-known as a PET tracer for imaging cellular proliferation in various cancers. The fact that this established tracer also appeared in the simulation results provides a strong validation of the QHPC–VS approach.
Moreover, the discovery of additional, previously unreported candidates opens new avenues for the design of imaging agents specifically targeting mutant IDH1. These molecules could form the basis for non-invasive diagnostic tools that improve the detection and monitoring of gliomas.
The success of this project was made possible by the immense computing power of JUWELS. The simulations required processing enormous numbers of calculations simultaneously, as quantum mechanical models involve tracking the electronic behavior of thousands of atoms at every time step. By distributing these calculations across JUWELS’ thousands of compute nodes, the MiMiC framework achieved excellent scaling—meaning that the simulation speed increased efficiently with more computing cores. This demonstrates that large-scale quantum-level simulations of biomolecules are now feasible, opening the door to routine use of QM/MM molecular dynamics in drug discovery.
Moreover, the project showcases how investment in national and European supercomputing infrastructure directly enables scientific advances with tangible biomedical applications. This research represents a significant step forward in how computational tools can be applied to drug design. By integrating quantum mechanics, high-performance computing, and automated workflows, the QHPC–VS protocol bridges the gap between theoretical chemistry and practical biomedical research. The implications extend far beyond the IDH1 enzyme or glioma imaging. Many other drug targets — such as metalloenzymes, covalent inhibitors, and transition-state analogs— involve complex electronic effects that classical simulations cannot capture. The same pipeline could therefore be adapted to study for example:
- Metal-dependent enzymes implicated in diseases ranging from cancers to
neurodegenerative diseases - Covalent inhibitors used in cancer therapy
- Enzyme mechanisms critical for diseases.
Furthermore, the combination of QM/MM and HPC could also improve our understanding of reaction mechanisms, drug metabolism, and toxicity, helping researchers design safer and more effective pharmaceuticals.
Until recently, quantum mechanical simulations were considered too expensive for practical applications in biology or pharmacology. The success of this work demonstrates that, with the right combination of algorithms, software frameworks, and HPC resources, quantum simulations can now contribute directly to real-world biomedical research. The MiMiC-based approach offers a scalable path to integrating QM/MM simulations into the standard drug discovery workflow. With supercomputers like JUWELS and the next-generation exascale systems now being developed across Europe, it will soon become feasible to include quantum accuracy in early-stage drug screening campaigns that examine thousands of molecules.