
MATERIALS SCIENCE AND CHEMISTRY
Understanding Doping Effects in Spin-Orbit Correlated Materials
Principal Investigator:
Prof. Dr. Eva Pavarini
Affiliation:
Forschungszentrum Jülich GmbH, Jülich, Germany
Local Project ID:
CTDMFTSO
HPC Platform used:
JUWELS CPU at JSC
Date published:
Abstract
Materials are made of electrons (negative charges) and nuclei (positive charges). The hydrogen atom is the simplest case: one nucleus and one electron. The behavior of a single electron attracted by a positively charged nucleus is complex, but also well understood: it is the quantum-mechanical version of a planet rotating around the sun. Materials contain many electrons, however. When the number of electrons increases the behavior of a system can radically change. This is because electrons strongly interact with each other: they are all negative charges and thus they try to avoid one another. When electron-electron repulsion effects dominate their behavior, electrons lose their individuality, forming cooperative emergent states. Understanding these emergent phases is one of the grand challenges in condensed-matter physics. Relativistic effects further complicate the matter. Progress can only be made exploiting the power of massively-parallel supercomputers via modern advanced computational techniques. The project CTDMFTSO follows this strategy, providing fundamental insight into these phenomena in paradigmatic classes of real materials.
Project Report
Many-body physics has the aim of explain the effects of the interactions among electrons in materials. This is known as the many-body problem.
Solving the many-body problem is one of the grand goals of condensed-matter physics. The difficulty arises from the fact that the behavior of one electron depends on the behavior of all others, and materials contain a very large number of electrons. In this situation unexpected emergent cooperative phenomena can arise. Examples are magnetic and orbitally ordered phases, metal-insulator transition and superconductivity. These phases are not only interesting from the fundamental physics viewpoint. They are in very important for novel technological applications; they can be exploited to reduce energy consumption, they are key for the developments of architectures that can enable quantum computation. Furthermore, understanding the many-body problem has implications beyond the world of quantum physics. The same type of emergent cooperative phenomena exists in all systems with interacting components, from elementary particles to biological systems, all the way to humans interacting in social media.
From the theory viewpoint, the problem in describing a system of interacting electrons is that conventional methods fail. This is because the latter assume that the interactions among the electrons just modify the shape of the landscape in which electrons move, while electrons still retain their individuality. This is an approximation that works well when the electron-electron repulsion is not the dominant effect, as it happens in certain categories of materials. However, when mutual interactions dominate, the effective landscape approaches break down qualitatively: electrons lose their individual character. It is thus necessary to resort to advanced computational techniques which explicitly account for this loss of individuality. There is catch, however: calculations become computationally extremely demanding. One can understand why in a simple manner: the behavior of one electron in a hilly landscape can be computed at the same computational cost of the behavior of one of an electron in a flat landscape. If, however, we have many electrons, the computational cost for describing their movement very rapidly increase, because to calculate what one electron does we have to know what all the others are doing at the same time: the landscape changes continuously. Understanding many-body effects in real materials thus requires exploiting the power of modern massively parallel supercomputing architectures via advanced computational many-body methods [1].
The project CTDMFTSO is based on one of the most successful many-body methods currently available for studying these phenomena in materials: the dynamical mean-field theory (DMFT) and its extensions. The goal of the project is to investigate relativistic effects; the latter give rise to exotic phases, anomalous superconductivity, entanglement phenomena and new form of ordering; at the same time, however, they make the challenge of simulating many-electron systems even harder. The project studies how relativistic effects change the strength of correlations. The latter is measured by the so-called effective mass. The concept of effective mass can be understood in a simple way. A free electron has a certain mass and correspondingly acquires a certain speed in an electric field. When electron interact together, however, the movement becomes slower because electrons scatter each other. One can describe this slow down as the electron acquiring an effective larger mass instead. Eventually, if interactions are very strong, the electron cannot move any more (the mass becomes infinitely large): a metal becomes insulator. The goal of the project CTDMFTSO was to study these effects in transition-metal oxides, a fundamental class of systems key for technological applications. Among the results obtained, the project could unravel the nature of correlations in iridates [2,3], a class of compounds that attracted a lot of attention because of their similarity with high-temperature superconducting cuprates. Calculations were made on the supercomputer JUWELS at the Forschungszentrum Jülich.
Results were disseminated via publications and invited talks at conferences as well as via the Autumn School on Correlated Electrons. The latter is a yearly event that brings at the FZJ each year key experts in the field as well as more than 100 early career scientists. The lectures of the Autumn School on Correlated Electrons can be found open access at www.cond-mat.de/events/correl.html. A book of lecture notes is published for each school. For the period in which CTDMFTSO was running, three schools were dedicated on topics very close to the project, as well as on massively parallel simulations of many-electron systems on supercomputers [4,5,6].
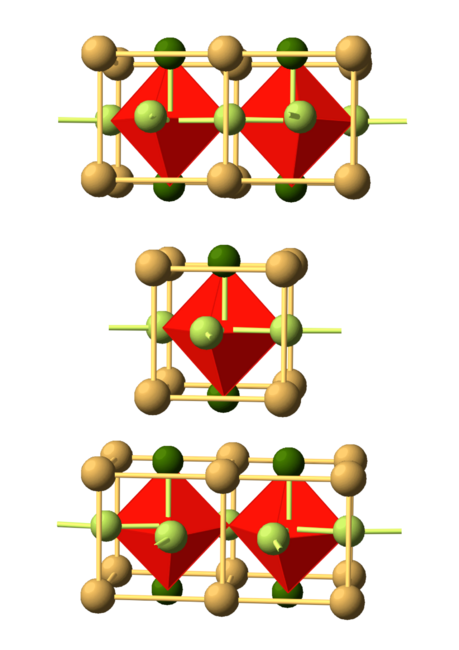
Figure: the crystal structure of Sr2IrO4
References and Links
[1] E. Pavarini, Rivista del Nuovo Cimento, Springer-Nature (2021)
[2] G. Zhang and E. Pavarini, Phys. Rev. Lett. 131, 036504 (2023)
[3] G. Zhang and E. Pavarini, Phys. Rev. B 104, 125116 (2021)
[4] Eva Pavarini and Erik Koch (eds.)
Topology, Entanglement, and Strong Correlations
Modeling and Simulation, Vol. 10
Verlag des Forschungszentrum Jülich, 2020
ISBN 978-3-95806-466-9
[5] Eva Pavarini and Erik Koch (eds.)
Simulating Correlations on Computers
Modeling and Simulation, Vol. 11
Verlag des Forschungszentrum Jülich, 2021
[6] Eva Pavarini, Erik Koch, Alexander Lichtenstein, and Dieter Vollhardt (eds.)
Dynamical Mean-Field Theory of Correlated Electrons
Modeling and Simulation, Vol. 12
Verlag des Forschungszentrum Jülich, 2022
ISBN 978-3-95806-619-9