
MATERIALS SCIENCE AND CHEMISTRY
Unveiling the Coordination Chemistry of Zinc(II) in a Protein: a QM/MM Study
Principal Investigator:
Dr. Davide Mandelli
Affiliation:
Forschungszentrum Jülich GmbH, INM-9 Institut für Neurowissenschaften und Medizin - Computational Biomedicine, Jülich, Germany
Local Project ID:
qmzinc
HPC Platform used:
JUWELS CPU at JSC
Date published:
Teaser Abstract
Zinc(II)-binding proteins play essential roles in biology, but their complex metal coordination is difficult to model accurately. Using an accurate Quantum Mechanics/Molecular Mechanics (QM/MM) molecular dynamics (MD) approach, this study explored the zinc(II) site of the Histone deacetylase protein, revealing detailed electronic and coordination dynamics. Leveraging our in-house MiMiC software on the JUWELS supercomputer, large-scale QM/MM MD simulations were performed efficiently providing insights into metal-ligand interactions that advance our understanding of metalloproteins.
Project Report
1. Introduction
Zinc(II)-binding proteins are critical for numerous biological functions, yet their intricate coordination environments pose challenges for classical simulation methods. Accurate modeling of these metal sites requires capturing electronic interactions that standard force fields cannot represent. This project focused on the zinc(II) binding site of the Histone deacetylase 6 (HDAC6) homodimer, employing a combined Quantum Mechanics/Molecular Mechanics (QM/MM) molecular dynamics (MD) approach to study coordination dynamics and electronic structure in detail. The computational complexity of QM/MM simulations necessitated the use of high-performance computing resources, with calculations performed on the JUWELS cluster.
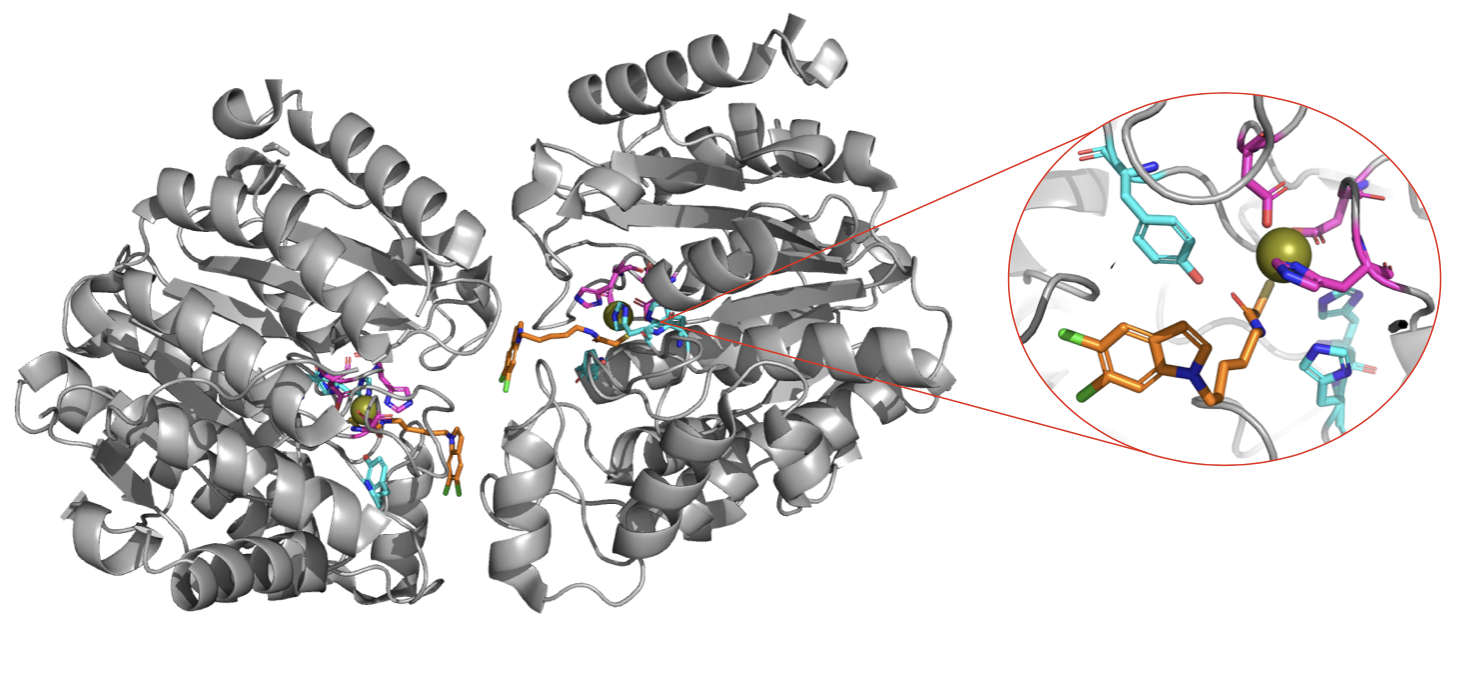
Figure 1. Protein and close up of the Zinc site in chain B of the homodimer. The zinc(II) ion (in gold) is coordinated by Asp 612, His 614 and Asp 705 (in pink). The metal is further interacting with the inhibitor (orange) which is stabilized by Tyr 745, His 573 and His 574 (cyan). In our QM/MM MD simulation, we considered the Zinc site in chain B as the QM region, while the rest of the system was treated classically.
2. Methods
a. Selection and Preparation of the System
The high-resolution crystallographic structure of HDAC6 (PDB ID: 6MR5) was selected. Each zinc(II) site features unique coordination, including a mercaptoacetamide inhibitor and nearby His and Asp residues (Figure 1). Preliminary classical MD simulations were performed using GROMACS [1] with the Amber99SBildn force field [2] for the protein and the nonbonded force field (NBFF) [3] for the zinc(II) site. The ligand was treated with the generalized AMBER force field (GAFF) [4]. The system, solvated with TIP3P water and neutralized with NaCl, underwent energy minimization, equilibration, and a 200 ns production run, confirming stable zinc(II) coordination.
b. QM/MM Simulations
Snapshots obtained from the preliminary classical MD were used to start the QM/MM simulations, where the QM region included the zinc metal ion, the inhibitor, and nearby stabilizing residues in chain B of the homodimer. Our highly scalable MiMiC framework (mimic-project.org) [5] was used for the QM/MM MD simulations. Specifically, we employed the Car-Parrinello MD approach, with the B3LYP functional [6], a plane-wave basis set, and Troullier-Martins pseudopotentials [7] used to set up the QM problem. Electrostatic interactions between QM and MM regions were computed using explicit short-range interactions and a seventh-order multipole expansion for long-range interactions [8]. All calculations were performed on JUWELS, utilizing 101 nodes: 100 for CPMD computations and one for GROMACS. Each node ran 8 tasks, with 6 CPUs per task.
3. Findings and Impact
Analysis focused on electron density, charge transfer, and polarization effects (see Figure 2). The presence of zinc(II) ion exerts a strong polarizing effect that significantly influences the surrounding residues. Charge analysis distribution within the coordination sphere reveals different interaction modes for each residue with the metal center. Upon binding, the NE2 lone pair of the histidine residue is substantially shared with the zinc(II) ion, resulting in a slight positive polarization on the donor atom. Similarly, the thiolate ligand, which is initially anionic, displays a reduction of the charge upon coordination. This reduction reflects strong metal–ligand polarization, with significant electron density transfer from sulfur to the zinc center. For zinc(II), a corresponding partial charge of q = 0.96 ± 0.01 is computed, indicating an effective gain of roughly 1 electrons relative to the formal +2 oxidation state. This value indicates pronounced polarization and effective electron transfer associated with coordination, reflecting significant electron delocalization.
The study demonstrated stable zinc(II) coordination and detailed electronic interactions within the HDAC6 binding site. Insights into metal-ligand interactions and charge redistribution enhance understanding of metalloprotein function, providing fundamental insights relevant for enzymology, drug design, and biotechnology in general. The methodology established here allows accurate modeling of complex metal-binding sites in proteins and can be extended to other metalloproteins with challenging coordination environments.
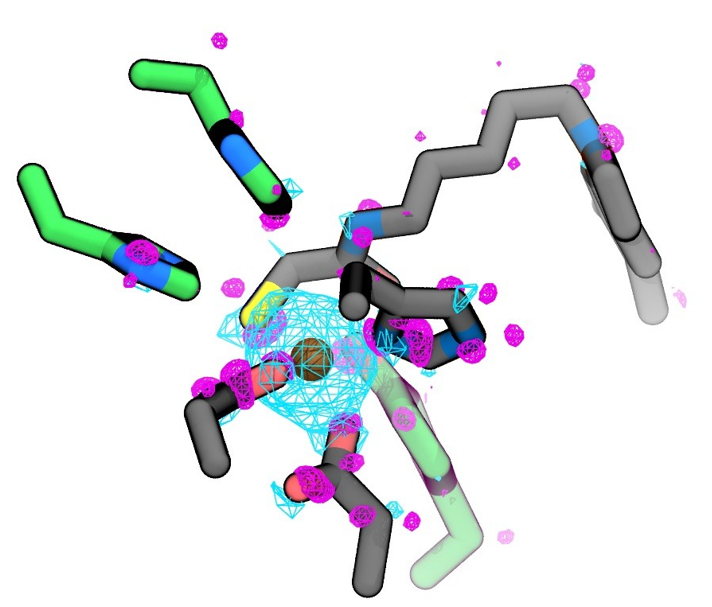
Figure 2. Difference in electron density Δρ=ρ^holo-ρ^apo between the presence and absence of Zn2+ for one QM/MM snapshot. Cyan and magenta surfaces represent an isovalue of +0.004 e / a03 and -0.004 e / a03 respectively. The residues coordinating the zinc(II) ion (in gold) are coloured in grey, while the residues stabilizing the inhibitor are coloured in green.
Outlook
Future work will extend simulation timescales and explore additional metalloprotein targets to refine QM/MM protocols further. The combination of high-accuracy simulations with HPC resources promises continued advances in understanding metal-dependent biological processes and guiding rational drug design.
References
[1] Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1-2, 19-25.
[2] Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. (2010). Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950-1958.
[3] Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. (2019). Upgrading and Validation of the AMBER Force Field for Histidine and Cysteine Zinc(II)-Binding Residues in Sites with
Four Protein Ligands. J. Chem. Inf. Model. 59, 3803-3816; Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. (2020). Upgraded AMBER Force Field for Zinc-Binding Residues and Ligands for Predicting Structural Properties and Binding Affinities in Zinc-Proteins. ACS Omega 5, 15301-15310.
[4] Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004). Development and testing of a general amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. doi.org/10.1002/jcc.20035
[5] Raghavan, B., Paulikat, M., Ahmad, K., Callea, L., Rizzi, A., Ippoliti, E., Mandelli, D., Bonati, L., De Vivo, M., & Carloni, P. (2023). Drug Design in the Exascale Era: A Perspective from Massively Parallel QM/MM Simulations. Journal of Chemical Information and Modeling, 63(12), 3647–3658. doi.org/10.1021/acs.jcim.3c00557
[6] Becke, A. D. (1992). Density-functional thermochemistry. II. The effect of the Perdew−Wang generalized-gradient correlation correction. J. Chem. Phys. 97, 9173−9177.
[7] Troullier, N., Martins, J. L. (1991). Efficient pseudopotentials for plane- wave calculations. Phys. Rev. B: Condens. Matter Mater. Phys. 43, 1993−2006.
[8] Olsen, J. M. H., Bolnykh, V., Meloni, S., Ippoliti, E., Bircher, M. P., Carloni, P., & Rothlisberger, U. (2019). MiMiC: A Novel Framework for Multiscale Modeling in Computational Chemistry. Journal of Chemical Theory and Computation, 15(6), 3810–3823. doi.org/10.1021/acs.jctc.9b00093