
ENVIRONMENT AND ENERGY
From Atoms to Eddies: Predictive-Quality Surface Reaction Chemistry in Real Reactor Models
Principal Investigator:
Christoph Scheurer
Affiliation:
Fakultät für Chemie, Technische Universtität München (Germany)
Local Project ID:
pr000aa
HPC Platform used:
SuperMUC of LRZ
Date published:
A team of scientists of the Technische Universtität München employed an instantaneous steady-state approximation to present steady-state reactivity data from kinetic Monte Carlo (kMC) simulations in the form of an interpolated data field as boundary conditions for the computational fluid dynamics simulation.Their goal was to test the capability of the code in managing complex computational domains, thus allowing for the first time to extend kMC simulations to geometries and conditions relevant to technological applications.
The worldwide rapidly growing demand for more efficient and sustainable exploitation of energy and material resources puts catalysis higher and higher up on the research agenda. New and improved catalysts are needed to deliver security of supply and industrial competitiveness. In particular, the rational design of new materials and processes requires the full and detailed understanding of the chemical reactor, which represents a key-component in many aspects of every-day life (from the production of important chemicals in large scale plants to the catalytic exhaust converters in cars).
In this respect, integrating first-principles kinetic Monte Carlo simulations (kMC) into the framework of computational fluid dynamics (CFD) is a crucial prerequisite. From a numerical point of view, a direct coupling between kMC and CFD is unfeasible especially for complex and realistic geometries, given the huge difference in characteristic times.
In order to overcome this, a team of scientists of the Technische Universtität München employed an instantaneous steady-state approximation to present steady-state reactivity data from kMC simulations in the form of an interpolated data field as boundary conditions for the CFD simulation. By means of this general methodology, the researchers integrated first-principles kMC simulations into the powerful CFD solver catalyticFoam for simulations of complex catalytic reactors [1]. Supercomputing power, such as provided by Petascale HPC system SuperMUC of the Leibniz Supercomputing Centre in Garching near Munich was crucial to test the capability of the code in managing complex computational domains (with millions of cells in the numerical domain), thus allowing for the first time to extend kMC simulations to geometries and conditions relevant to technological applications.
By the analysis of several geometries and real conditions, the scientists demonstrated the intricate coupling of the reactive surface chemistry and flow that significantly modifies the observable catalytic function. On the whole, the developed approach establishes an accurate, well formalised, and fully integrated multiscale framework that roots in a first-principles description of the catalyst and includes all larger-scale aspects up to the full reactor in an ideal seamless flow from atoms to eddies [2]. Such a tool will be of direct use for the development of new reactor and reaction concepts for advanced energy and chemical applications.
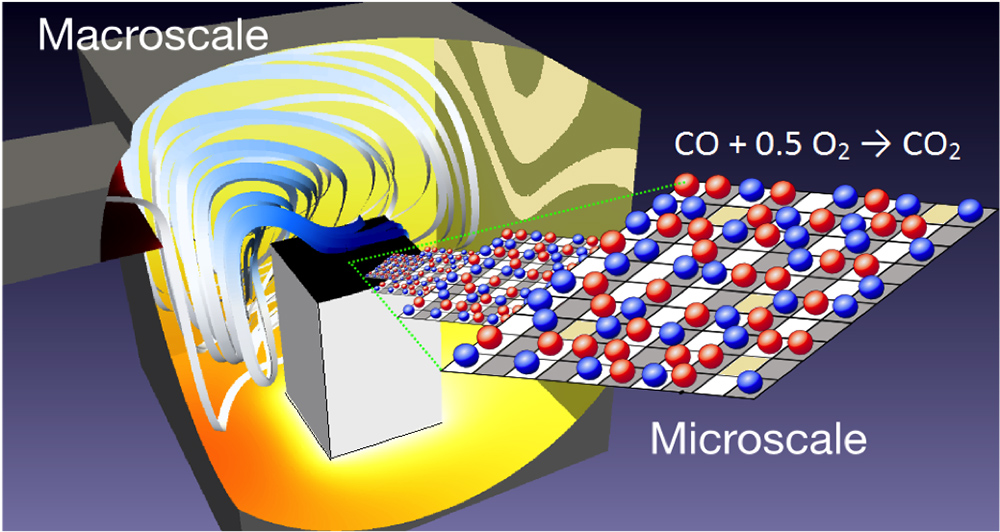
Fig. 1. Representation of a reactor geometry considered as prototypical for an in-situ spectroscopic cell.
Copyright: Technische Universität München, Fakultät für ChemieReferences:
[1] M. Maestri, A. Cuoci, catalyticFoam, (2011-2014), www.catalyticfoam.polimi.it
[2] S. Matera, M. Maestri, A. Cuoci, K. Reuter, Predictive-quality surface reaction chemistry in real reactor models: Integrating first-principles kinetic Monte Carlo simulations into computational fluid dynamics, ACS Catalysis, 4 (2014) 4081–4092
Scientific contact:
Dr. Christoph Scheurer
Technische Universtität München, Fakultät für Chemie
Lichtenbergstraße 4
D-85748 Garching/Germany
e-mail: christoph.scheurer@ch.tum.de