
MATERIALS SCIENCE AND CHEMISTRY
2D Material Growth on Liquid Metals: Role of Liquid Copper in High-quality Graphene Synthesis
Principal Investigator:
Karsten Reuter
Affiliation:
Lehrstuhl für Theoretische Chemie, Technische Universität München
Local Project ID:
LMcat
HPC Platform used:
JUWELS of JSC
Date published:
Introduction
Graphene is a 2D material made up of a single atomic layer of carbon atoms. It finds applications in many technological areas owing to its remarkable structural and electronic properties such as its strength and flexibility and the exceptionally high electrical conductivity. However, a full exploitation of these properties requires the development of mass production techniques. Furthermore, control of the level of defects and impurities is crucial since the properties of graphene are highly sensitive to any deviations from the crystalline lattice.
Within the last decade, liquid Cu has emerged as an interesting catalyst for fast and efficient industrial-scale production of high-quality graphene. However, the current knowledge about the catalytic properties of liquid metal catalysts is extremely poor, as they had no technological significance in the past. Together with experimental partners from The Netherlands, France and Greece, researchers from Technical University of Munich are part of a European project (LMCat) that aims to unravel the growth mechanism of graphene on liquid Cu by combining experiments and computer simulations.
From the experimental side, a novel reactor that allows real-time monitoring and control of the growth via in situ optical microscopy, synchrotron X-ray diffraction, and Raman spectroscopy has been constructed. An example of such in situ monitoring of the dynamics and self-assembly of graphene flakes using optical microscopy is shown in Figure 1. The computer simulations were carried out on the JUWELS Cluster in Jülich and focused on studying the molecular building blocks for graphene growth and on developing an understanding of the reaction mechanism and the mesoscopic self-assembly of the flakes observed experimentally. Such insight is important in order to describe and optimize 2D material growth at liquid metal catalysts.

Figure 1. In situ optical microscopy image of self-assembled graphene flakes grown in the LMCat reactor. Picture courtesy of: Maciej Jankowski, CEA / Institute for Nanoscience and Cryogenics, Grenoble, France.
The Challenge
Theoretical approaches to elucidate the mechanism of 2D material growth on liquid metal catalysts are challenged by the need to account for the highly dynamic properties of the liquid. Even using modern day supercomputers such as the JUWELS Cluster, dynamical simulations based on quantum mechanical methods such as density functional theory (ab initio molecular dynamics, AIMD) are exceedingly expensive for the required system sizes and can only probe simulation times on the order of picoseconds. To overcome this challenge, various methods were combined as discussed in the following section.
Results
In the first part of the project ab initio thermodynamics based on static density functional theory calculations was used to assess the stability of a wide range of hydrocarbons under typical reaction conditions [1]. These results showed that the reaction precursor (methane) is likely to lose all of its hydrogen atoms to the gas phase, leaving only pure carbon synthesis building blocks at the surface. Short timescale AIMD of smaller graphene flakes containing only carbon atoms were then carried out and used to benchmark the performance of a cheaper empirical description of the system using the COMB3 reactive force field. In particular, evidence that the flakes may embed into the liquid surface (see Figure 2) was found using both levels of theory [2].

Figure 2. Snapshot from dynamical simulation of a graphene flake on liquid copper showing the embedded state of the flake. © TUM
With the cheaper force field description, it became possible to simulate graphene flakes of various sizes for much longer timescales (~1 ns) and to derive input parameters for a mesoscopic model of the system (see Figure 3A) [3]. This model was used to explain the experimental observation of a coalescence of the growing hexagonal flakes into a close-packed structure with well-defined inter-flake separation (Figure 1) in terms of long-range capillary and electrostatic interactions (Figure 3B). Figure 3C shows the dependence of the optimum interflake distance on the model parameters and demonstrates a good qualitative agreement with the experimentally observed distance of 40 μm. Figure 3C shows the dependence of the optimum interflake distance on the model parameters and demonstrates a good qualitative agreement with the experimentally observed distance of 40 μm.
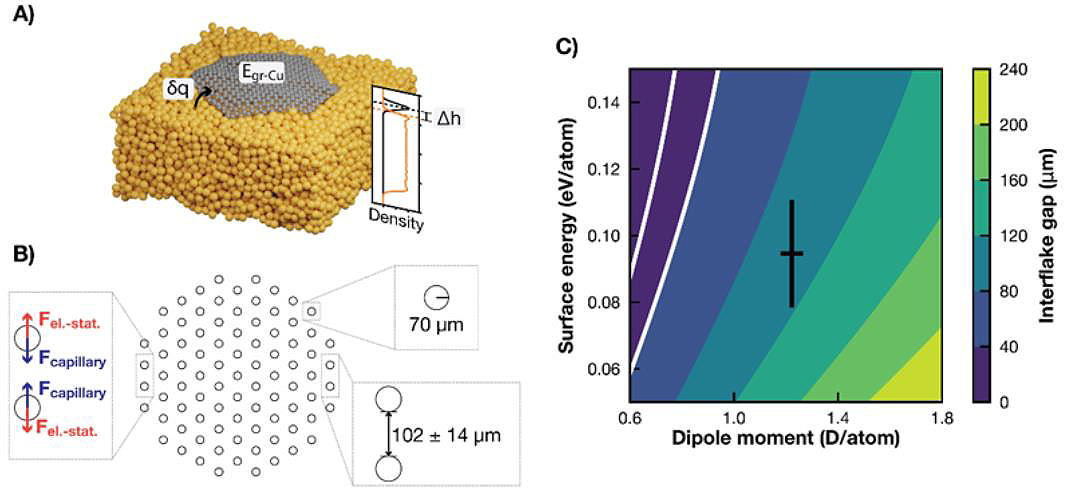
Figure 3. A) Illustration of properties derived from the dynamical simulations, namely the charge transfer from the liquid Cu to the flake, δq, the interaction energy between graphene and copper, Egr-Cu, the height of the flake above the surface, Δh, as well as the accompanying density profile. The orange line corresponds to the density of Cu atoms within the hexagon’s inscribed circle and the black line to the C atomic density. The densities have been arbitrarily scaled for better visualization. B) Schematic representation of the group of interacting flakes used to calculate the separation distance. The insets indicate the two governing forces, the used flake radius, and the optimized distance. C) Average distance between flakes as predicted by the capillary-electrostatic model as a function of the two model parameters. The black cross corresponds to the dipole moment and interaction energy as obtained from our MD simulations with their corresponding standard deviations. The white lines denote 20 and 40 μm gap distances (area in agreement with the experiments). Reproduced from Ref. [3].
Who might be interested in these results?
The catalytic properties of liquid metals are still largely unknown territory. The results obtained in this LMCat project begin to probe some of the fundamental questions how molecules and larger flakes of 2D materials such as graphene interact with a liquid surface, as well as how interactions between the flakes themselves are mediated by charge transfer to or from the liquid and deformations of the liquid, the latter leading to capillary interactions. Such understanding of the growth dynamics and the forces acting between flakes at a liquid metal surface is important for further tailoring the growth conditions in order to control the quality of the final graphene product.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement 736299. Responsibility for the information and views set out in this report lies entirely with the authors.
References and Further Links
[1] Mie Andersen, Juan Santiago Cingolani, Karsten Reuter. J. Phys. Chem. C 123, 22299 (2019). pubs.acs.org/doi/abs/10.1021/acs.jpcc.9b05642
[2] Juan Santiago Cingolani, Martin Deimel, Simone Köcher, Christoph Scheurer, Karsten Reuter, Mie Andersen. J. Chem. Phys. 153, 074702 (2020). aip.scitation.org/doi/abs/10.1063/5.0020126
[3] Maciej Jankowski, Mehdi Saedi, Francesco La Porta, Anastasios Manikas, Christos Tsakonas, Juan Santiago Cingolani, Mie Andersen, Marc de Voogd, Gertjan van Baarle, Karsten Reuter, Costas Galiotis, Gilles Renaud, Oleg Konovalov, Irene Groot (in preparation).
https://www.youtube.com/watch?v=sp6VujNRDRI
https://www.youtube.com/watch?v=ryRFu9yhHUU&feature=emb_logo
Research Team
Mie Andersen, Juan Santiago Cingolani, Karsten Reuter (PI)
All: Lehrstuhl für Theoretische Chemie, Technische Universität München, Germany
Scientific Contact
Dr. Mie Andersen
Theoretische Chemie, Technische Universität München
Lichtenbergstraße 4, D-85747 Garching/Munich (Germany)
e-mail: mie.andersen [at] ch.tum.de
http://www.th4.ch.tum.de/andersen
Local project ID: LMcat
February 2021