
MATERIALS SCIENCE AND CHEMISTRY
Ultrafast Excited-State Relaxation Dynamics of Proton and Solvated Electron in Liquid Water
Principal Investigator:
Prof. Dr. Thomas Bredow
Affiliation:
Universität Bonn (Germany)
Local Project ID:
hbn34
HPC Platform used:
JUQUEEN of JSC
Date published:
Ab initio calculations are carried out to study chemical processes and relaxation dynamics of water in its excited states upon photo-excitation. In this project, the researchers discovered an unusual non-grotthus-like proton transfer and a mixed localized and enhanced spin density distribution of solvated electron in water using combined Born-Oppenheimer molecular dynamics and time dependent density functional theory within periodic boundary condition. These investigations led to a deeper understanding of ultra-fast excited-state processes in fluids and are of general importance for physical chemistry of excited-state phenomena.
Introduction
By the advent of femtosecond chemistry real-time tracking of ultra-fast processes in excited states of chemical systems became accessible, allowing for exploring a rich scientific area of unprecedented chemical and physical importance. In this regard, one of the most intense studied systems in almost every aspect is liquid water; however its excited-state dynamics in condensed phase has been much less matter of theoretical investigations. The experimental measurements confirm occurrence of non-trivial excited-state processes at low and high energies. The vertical transition energy of the bulk liquid water from valence to conduction band continuum is reported at 11.1 eV at ambient temperature1,2, indicating that at this energy and above, the initially non-thermal conduction band electron and the cationic hole are solely formed by a direct photo-ionization process:
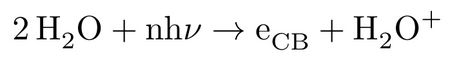
followed by further relaxation of the highly unstable H2O+ upon a proton transfer (PT) to a nearby water molecule according to:
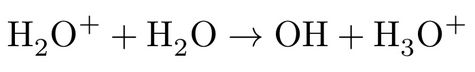
resulting finally in a thermalized hydrated electron, a hydronium cation and a hydroxyl radical. However, the most striking point from excitation perspective is that such above-band-gap process with exactly the same final net products have also been observed experimentally at energies far lower than the electronic band gap, extending to the optical absorption band edge of water3-5, showing that below-band gap processes are possible only through an explicit involvement of solvent nuclear motions6. Currently two mechanisms are debated as the most leading candidates for below-band gap excitations, namely, proton-coupled electron transfer (PCET) and hot hydrogen atom (HHA) mechanism7-9,11,12 .
PCET involves a proton transfer along the proton coordinate to a neighbouring water molecule, as well as injection of an electron into a presolvated solvent trap, stabilizing the excess charge, i.e.:
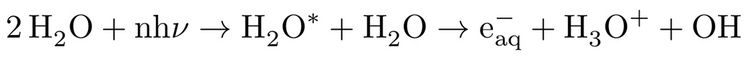
while, in HHA process, due to dissociative character of the excited states of water11,13, a translationally hot hydrogen atom according to: H2O* --> H (hot) + OH with high kinetic energy profile is ejected which subsequently collides with a water molecule from its surrounding, leading to formation of a hydrated electron, hydronium cation and hydroxyl radical, i.e.:
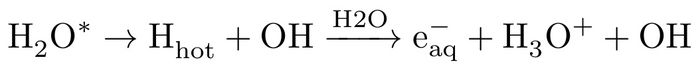
Both low energy pathways (representing indirect ionisations) need a significant change of water solvent network (to produce the hydrated electron), serving as an important criterion for feasibility of the reactions. In both reactions (3 and 4) the same net products are generated on different pathways.
Computations
In order to verify which one of the reaction candidates (3 or 4) is the most probable one, the researchers analysed low energy excited-state dynamics of liquid water by means of ab-initio time-dependent density functional theory combined with ab-initio Born-Oppenheimer molecular dynamics14-16 under periodic boundary condition with plane waves to avoid surface effects. However, since the relaxation dynamics needs to be sampled for many water snapshots, modelled within a large simulation box (to avoid finite size effects), the ab-initio excited-state calculations require massive computation power, and therefore, JUQUEEN provides an optimal platform to realize this research.
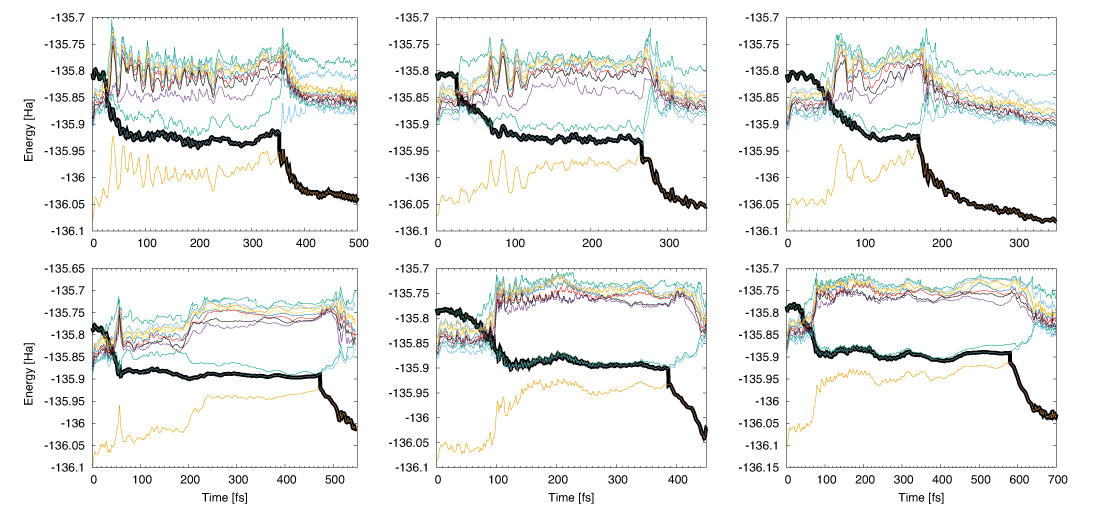
Fig. 01: Non-adiabatic dynamics of water for 3 independent initial configurations from S10 to S0(ground state) using PBE in the upper and PBE0 functional in the lower panel. The consecutive hoppings to the lower states were found through explicit ab-initio computation of non-adiabatic couplings at each step of excited-state dynamics and tracked by the thick black line (the running state). The black line describes how state hopping from higher to lower states at what times occurs. The other colours describes the dynamics in the corresponding excited states if hopping is not taken into account. The first yellow line describes the ground-state dynamics (S0). Depending on the initial snapshots the rest time of the excited-state dynamics on S1 varies between 100-250 fs for the PBE and 300-500 fs for the PBE0 functional, respectively.
Copyright: THCH, University of BonnResults
In Fig. 1 the non-adiabatic dynamics of water starting from S10 (a singlet state which lies nearly 1 eV above the S1) for PBE and PBE0 functionals is presented. The researches observed consecutive fast hoppings from S10 to S1 within some femtoseconds, but with a relatively long rest of the system in S1 of about 100 to 500 fs before relaxation to S0 is achieved. Variation of the rest time of S1 depends on the initial configurations and more importantly on the approach. The PBE0 functional enhances the S1 rest lifetime, whereas PBE relaxes the system from S1to the ground-state S0 in a shorter time; however both functionals lead to similar results, such as fast initial consecutive relaxations and a relatively long period of rest time in S1 and hence an ultra-fast transfer of proton to the neighbouring molecules. This underlines the fact that proton transfer is independent of the description of the exchange-correlation potential, and the observed complex dynamics of proton indeed stems from the intrinsic physics of the system.
Based on excited-state dynamics for many water snapshots following possibilities are observed:
i) upon photo-excitation of a single water molecule (surrounded by a solvent shell) a fast proton transfer to the next nearby water molecule is occurred (12 fs) followed by a localization of about 53 fs, after which a full PT to the third water molecule is accomplished (76 fs) on which it gets delocalized over a water wire for a very short period of time (92 fs), and again localizes on the third water molecule, followed by short delocalisations on the nearby lying forth water molecule (128 fs).
ii) after a rapid PT to the neighbouring water molecule (8 fs), the proton rests on it for a relatively long period of time of about 106 fs, followed by forward and backward hops between the second and third molecule, till finally a full PT to the third molecule is accomplished (191 fs), from which it can further jump to the neighbouring molecules
iii) after the initial excitation an instantaneous PT from the first to the second and from the second to the third water molecule occurs within less than 11 fs. Once the proton reaches the third molecule, after a quick while it gets delocalized over the second and third molecule for a short time, after which it again becomes localized on the third molecule. However, due to sudden creation of a water wire, it delocalizes over 4 water molecules for a short time (56 fs). After this massive delocalization, it again localizes on the third molecule, from which consecutive hops to the fourth and fifth molecules occur (114 fs). This is shown explicitly in Fig. 2.
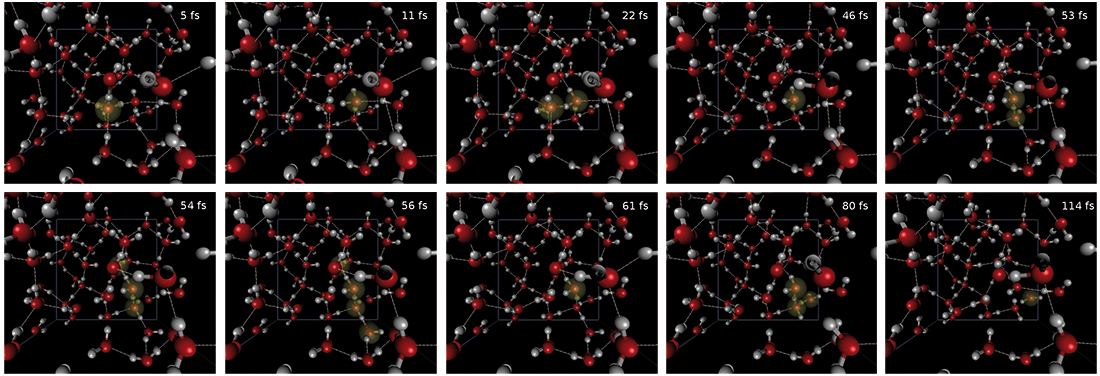
Fig. 02: The picture shows multiple forward, backward, and delocalisations of a proton upon excitation. The protonated water is indicated by the yellow sphere.
Copyright: THCH, University of Bonniv) breaking of water bonds followed by fast recombination. Thus, after the initial excitation, a solvated oxygen atom can be formed which again rapidly recombines with its protons, after which a discrete PT follows as described above.
A further interesting point is that although proton dynamics shows multiple back and forth transitions with the possibility of getting delocalized over some neighbouring water molecules or getting stuck on a water molecule for a relatively long period of time due to a defected hydrogen bond network, the hydroxyl radical (OH) however exhibits an extremely low diffusion and is non-reactive during the entire excited-state run.
Concerning solvated electron density, as shown in Fig. 3, the solvated electron is to a large extent localized in a particular region of space (far from the remaining OH radical which is indicated by the yellow colour) within the first solvent shell with some notable overlappings with the nearby water molecules and delocalizing tails extending the first and second solvent sphere. This indicates that he spatial extent of the solvated electron is made of a mixture of three densities; a localized part within a cavity, which contains the largest part of the total spin density, an enhanced density area on the water molecules, and a delocalizing density (in form of diffusive tails) which is the smaller part and distributed anisotropically mostly within the second and (to a lesser extent) third solvent sphere. The delocalisations beyond the cavity are likely artifacts of the underlying density functional. The observed enhanced density on the nearby water molecules points at sizable overlap of electron density with the surrounding water molecules. This complex picture of spin distribution indicates at a mixed repulsive and attractive interaction between the solvated electron and the solvent shell17-20. Consequently, the true spin distribution is highly sensitive to the details and interplay of repulsive and attractive interactions which can not be perfectly captured by density functional theory (DFT) due to its approximative nature.
These large-scale excited-state calculations show that instantaneous consecutive hops of proton to the neighbouring water molecules (grotthuss mechanism) is highly unlikely, and the ejected solvated electron is not fully localized within a cavity-like environment due to attractive interaction with the immediate surrounding water molecules.
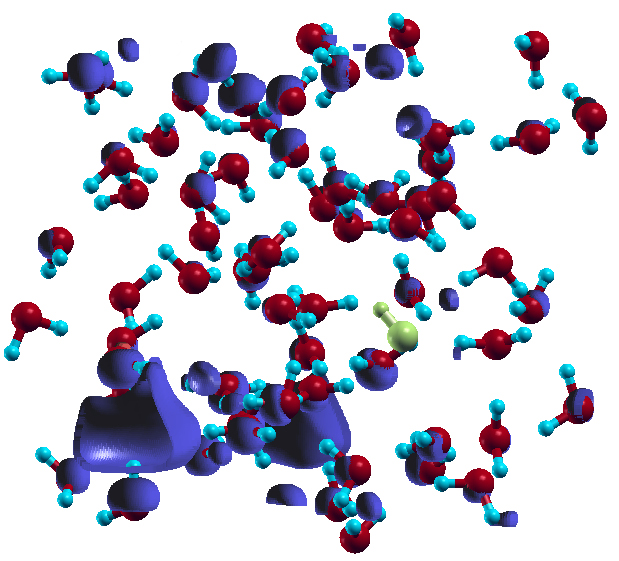
Fig. 03: Spin density of the solvated electron (shown in blue) at maximum propagation distance of proton (after 114 fs) from the OH radical indicated by the yellow color in the box. The localized density coordinated to six water molecules can be seen in bottom left and in the background (periodic boundary condition). The overlapping density on the water molecules around the localized part is obvious. The diffusive tails appear most in the upper half of the box.
Copyright: THCH, University of BonnAcknowledgement
The researchers gratefully thank Jülich Supercomputing Centre for providing computing time for the project HBN341 and the Deutsche Forschungsgemeinschaft for financial support within the Collaborative Research Center SFB 813: Chemistry at Spin Centers, Project A2, Theoretical methods for studying long-range charge and spin separation in excited states of large molecules, solids and surfaces.
References:
[1] Elles, C. G.; Jailaubekov, A. E.; Crowell, R. A.; Bradforth, S. E. J. Chem. Phys. 125, 044515 (2016).
[2] Winter, B.; Weber, R.; Widdra, W.; Dittmar, M.; Faubel, M.; Hertel, I. V. J. Phys. Chem. A 108 , 2625 (108).
[3] Bernas, A.; Ferradini, C.; Jay-Gerin, J.-P. J. Photochem. Photobiol. A 117 , 171 (1998).
[4] Marsalek, O.; Uhlig, F.; VandeVondele, J.; Jungwirth, P. Acc. Chem. Res. 45, 23 (2012).
[5] Marin, T. W.; Takahashi, K.; Bartels, D. M. 125 , 104314 (2006).
[6] Coe, J. V.; Earhart, A. D.; Cohen, M. H.; Ho_man, G. J.; Sarkas, H. W.; Bowen, K. H. J. Chem. Phys. 107, 6023 (1997).
[7] Kratz, S.; Torres-Alacan, J.; Urbanek, J.; Lindner, J.; Vohringer, P. Phys. Chem. Chem. Phys. 12, 12169 (2010).
[8] Crowell, R. A.; Bartels, D. M. J. Phys. Chem. 100, 17940 (1996).
[9] Thomsen, C. L.; Madsen, D.; Keiding, S. R.; Thogersen, J.; Christiansen, O. J. Chem. Phys. 110, 3453 (1999).
[10] A. Hassanali et al. PNAS, 110, 13723 (2013).
[11] Elles, C. G.; Shkrob, I. A.; Crowell, R. A.; Bradforth, S. E. J. Chem. Phys. 126, 164503 (2007).
[12] Torres-Alacan, J.; Kratz, S.; Vohringer, P. Phys. Chem. Chem. Phys. 13, 20806 (2011).
[13] Engel, V.; Schinke, R.; Staemmler, V. J. Chem. Phys. 88, 129 (1998).
[14] E. Tapavicza, I. Tavernelli, and U. Rothlisberger, Phys. Rev. Lett. 98, 023001 (2007).
[15] E. Tapavicza, I. Tavernelli, U. Rothlisberger, C. Filippi, and M. E. Casida, J. Chem. Phys. 129, 124108 (2008).
[16] I. Tavernelli, B. F. E. Curchod, A. Laktionov, and U. Rothlisberger, J. Chem. Phys. 133, 194104 (2010).
[17] R. E. Larsen, W. J. Glover, B. J. Schwartz, Science 329, 5987 (2010).
[18] L. D. Jacobson, and J. M. Herbert, Science 331, 6023 (2011).
[19] J. R. Casey, A. Kahros, and B. J. Schwartz, J. Phys. Chem. B 117, (46), pp 1417314182 (2013).
[20] F. Uhlig, O. Marsalek, and P. Jungwirth, J. Phys. Chem. Lett. 3, (20), pp 3071-3075 (2012).
Research Team:
Vafa Ziaei and Prof. Dr. Thomas Bredow (PI), Mulliken Center for Theoretical Chemistry, University of Bonn, Bonn, Germany
Scientific Contact:
Vafa Ziaei
Mulliken Center for Theoretical Chemistry
University of Bonn
Beringstr. 4, D-53115 Bonn (Germany)
E-mail: ziaei [@] thch.uni-bonn.de
June 2017