
LIFE SCIENCES
Statistical Inference for Large-scale Ordinary Differential Equation (ODE) Models of Cancer Signalling
Principal Investigator:
Jan Hasenauer
Affiliation:
(1)Institute of Computational Biology, Helmholtz Zentrum München - German Research Center for Environmental Health, (2)Center for Mathematics, Technische Universität München, (3)Faculty of Mathematics and Natural Sciences, University of Bonn
Local Project ID:
pr62li
HPC Platform used:
SuperMUC and SuperMUC-NG of LRZ
Date published:
Mechanistic computational modelling using ODEs (ordinary differential equations) is widely used in systems biology [1]. These ODE models allow for the integration of heterogeneous data with prior information to provide a quantitative understanding of complex biological systems. Parameters of mechanistic models are often unknown and need to be inferred from experimental data. The need for repeated numerical simulation and gradient evaluation renders this statistical inference computationally very demanding. Therefore, current models are usually restricted to very small subsets of cellular processes which cannot account for many aspects underlying complex diseases such as cancer.
Especially in the context of cancer, there are large drug screening and genome and transcriptome sequencing data sets available [2,3] which are generally analysed using purely statistical approaches. Recent methodological developments enabled the parameterization of a large mechanistic model describing multiple cancer-relevant cellular signalling pathways [4]. Fröhlich et al. showed that parameterized mechanistic models generalize as well as statistical models, but additionally provided information on latent variables as well as a more mechanistic understanding of the underlying process.
To allow for the integration of even larger datasets which are expected to further improve model generalization, researchers from the Helmholtz Zentrum Munich set out to evaluate current, and to develop novel, more efficient parameter estimation algorithms.
Results and Methods
Scalable model simulation and parameter inference
This work was based on a recently published large-scale ODE model describing molecular signal processing in multiple cancer cell lines [4]. The model contains over 4000 kinetic parameters to describe the dynamics of over 1000 state variables such as proteins, protein complexes and drugs (Figure 1). Model parameters were estimated based on published data from a large number of drug response viability assays and basal (phospho-)protein measurements [2,5]. Existing gradient-based non-linear optimization methods were used for maximum likelihood estimation.
Multi-start local optimization was performed using a custom open source C++/MPI-based library parPE [6]. For the numerical simulation of the ODE model the open source software AMICI [7] was used, which builds upon Sundials solver suite [8]. The scalable adjoint sensitivity method was applied to compute gradients for optimization. ODE simulations for different experimental conditions were run in parallel for each objective function evaluation. Jobs were run on SuperMUC Phase 2 thin nodes using up to 128 nodes for 25 parallel local optimizations or on 12 nodes for individual local optimizations. Computation time for a single optimization run of 150 iterations was about 4000 core hours. The total compute time budget spent during this project was over 6 million core hours.

Figure 1: Schematic overview of mechanistic pan-cancer model. The model comprises protein synthesis, protein-protein and protein-drug interaction and cell proliferation. It can account for 36 cancer-related activating mutations. See [4] for more details. (Figure from [4] used under CC BY-NC-ND 4.0 / figure cropped).
Evaluation of existing optimizers and development of an efficient hierarchical optimization approach
The granted computation time on SuperMUC allowed for a performance evaluation of different existing local optimization algorithms, such as Ipopt [9] and Ceres [10]. Although in the chosen setting Ipopt was found to perform best (Figure 2), it became apparent that all tested optimizers had trouble handling the relative measurements used for model calibration. In particular, none of the tested optimization routines achieved a reasonable fit within the allowed computational budget.
Being faced with the unsatisfactory optimizer performance lead Schmiester et al. [11] to the development of a more efficient hierarchical optimization approach. This approach allows for splitting the overall optimization problem into two smaller ones, the inner of which can often be solved analytically. Evaluating this approach in combination with various optimizers showed drastic improvements in optimizer performance (Figure 2).

Figure 2: Application of the developed hierarchical optimization approach with different optimizers (Figure from [11] used under CC BY 4.0). (A) Optimizer trajectories of the standard and the hierarchical optimization for four different optimizers. (B) The final objective function values found by the different approaches are compared. (C) Speed up factors using the hierarchical optimization as compared to the standard approach.
Mini-batch optimization
For the chosen dataset, a single cost function evaluation during optimization required the numerical simulation of thousands of large ODE systems, adding up to thousands of hours of computation time for the full optimization. In the field of deep learning, where models need to be evaluated on a much larger number of datasets, mini-batch optimization is commonly used [12]. The basic idea is that objective function is evaluated not on the full dataset, but only on a subset thereof. This approach allows for more frequent parameter updates during optimization at the cost of stochastic gradient approximations.
The granted computational budget enabled Stapor et al. [13] to connect efficient solvers for dynamical systems with methods from the field of deep learning. They evaluated state-of-the-art mini-batch optimizers and extended them to improve robustness for solving parameter estimation problems for ODE models. These novel algorithms allowed for a several-fold speed up as compared to previous approaches (Figure 3).
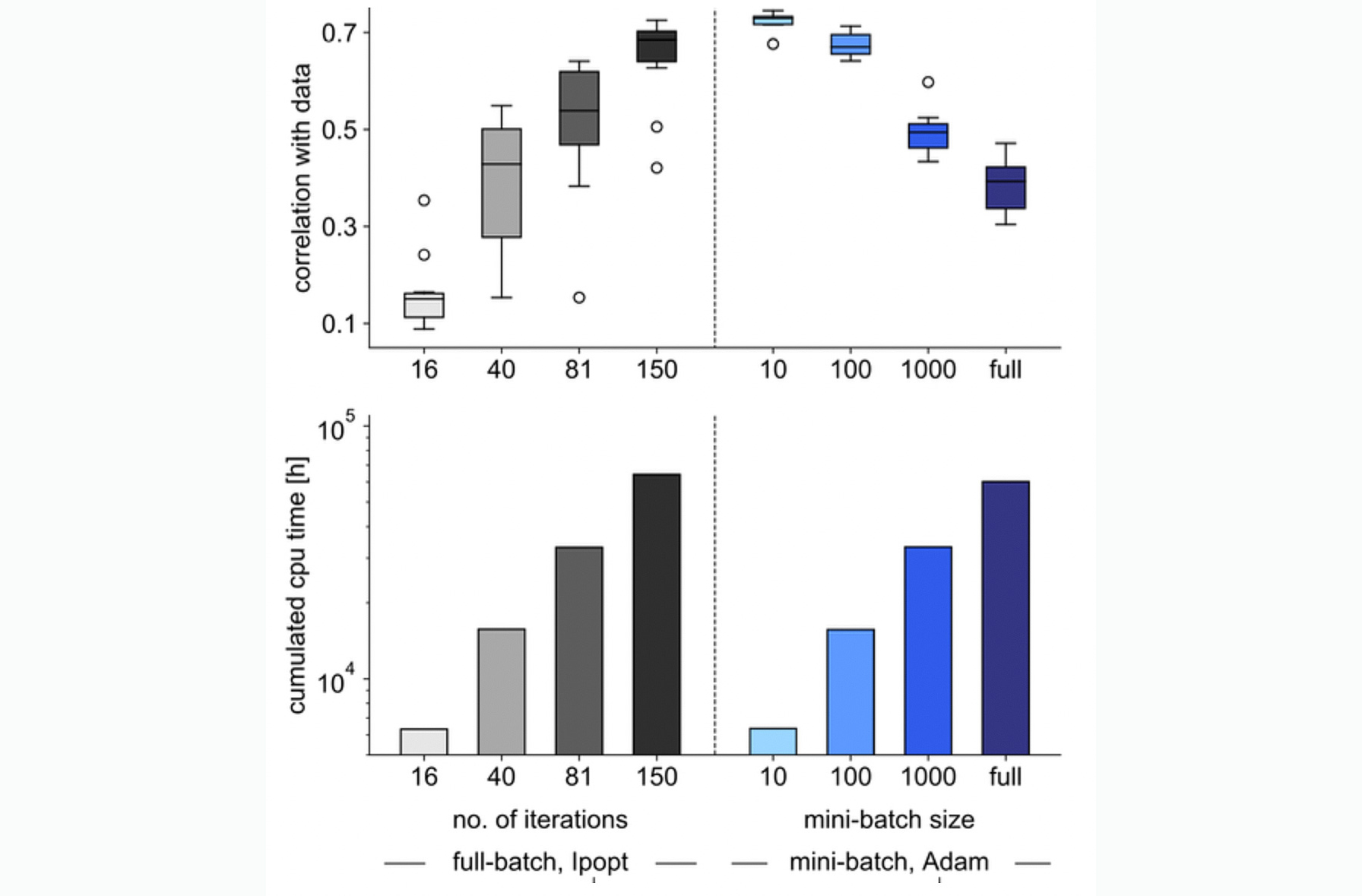
Figure 3: Mini-batch optimizers allow for more efficient model calibration than batch optimizers. Comparison of optimization results for a large-scale cancer model with a full batch optimizer and a mini-batch optimizer with different mini-batch sizes (20 optimizer runs, both algorithms started at the same random parameters), for Ipopt at four different stages of the optimization process to compare performance over computation time and for mini-batch optimization with different mini-batch sizes. Top: Correlation of model simulation with measurement data (best 10 runs). Bottom: Total computation time for all 20 optimization runs. For details see [13]. (Figure from [13] used under CC BY 4.0)
On-going Research / Outlook
The computing time granted on SuperMUC enabled the researchers to evaluate gradient-based batch and mini-batch optimizers for parameter estimation for large-scale mechanistic models. Based on these results, a novel hierarchical optimization approach was developed for use with adjoint sensitivity analysis which drastically improved optimizer convergence in the presence of relative data.
The obtained results will be of interest to the systems biology community and will feed directly into the Horizon 2020 project CanPathPro [14] which focuses on predictive modelling in the cancer context.
After having made significant methodological progress, the researchers plan on applying these tools in a more biology-driven follow up project focusing on model selection and integration of additional data types.
Researchers
Principal Investigator: Jan Hasenauer1,2,3
Researchers: Daniel Weindl1, Leonard Schmiester1,2, Paul Stapor1,2, Yannik Schälte1,2
Affiliations:
1 Institute of Computational Biology, Helmholtz Zentrum München -- German Research Center for Environmental Health, 85764 Neuherberg, Germany
2 Center for Mathematics, Technische Universität München, 85748 Garching, Germany
3 Faculty of Mathematics and Natural Sciences, University of Bonn, 53113 Bonn, Germany
Acknowledgement
This work was supported by the German Research Foundation (Grant No. HA7376/1-1; Y.S.), the German Federal Ministry of Education and Research (SYS-Stomach; Grant No. 01ZX1310B; J.H.), and the European Union’s Horizon 2020 research and innovation program (CanPathPro;Grant No. 686282; P.S., J.H., D.W.). Computer resources for this project have been provided by the Gauss Centre for Supercomputing / Leibniz Supercomputing Centre under grant pr62li.
References and Links
[1] Ingalls, BP. Mathematical modeling in systems biology: an introduction. MIT press, 2013.
[2] Barretina, J. et al., Nature, 483, 7391, 603–607, 2012; https://doi.org/10.1038/nature11003
[3] Yang W. et al., Nucl. Acids Res., 41, D955–D961, 2013; https://doi.org/10.1093/nar/gks1111
[4] Fröhlich, F. et al., Cell Systems 7, 6, 2018, 567-579.e6; https://doi.org/10.1016/j.cels.2018.10.013
[5] Li, J. et al., Cancer cell, 31, 2, 225–239, 2017; https://doi.org/10.1016/j.ccell.2017.01.005
[6] Weindl, D. et al., parPE parameter estimation library, https://github.com/ICB-DCM/parPE/
[7] Fröhlich, F. et al., Advanced Multi-language Interface to CVODES and IDAS (AMICI) https://github.com/ICB-DCM/AMICI/
[8] Hindmarsh, A. C. et al., ACM T. Math. Software., 31, 3, 363–396, 2005; https://doi.org/10.1145/1089014.1089020
[9] Wächter, A. and Biegler L., Mathematical Programming, 106, 1, 25–57, 2006; https://doi.org/10.1007/s10107-004-0559-y
[10] Agarwal, S. et al., Ceres Solver, http://ceres-solver.org
[11] Schmiester, L. et al., Bioinformatics, btz581; https://doi.org/10.1093/bioinformatics/btz581
[12] LeCun Y. and Bottou L., Lecture Notes in Computer Science, vol. 1524, Orr and Müller, Eds. Springer Berlin Heidelberg, 2002, pp. 9–50.
[13] Stapor, P. et al., bioRxiv, 859884, 2019; https://doi.org/10.1101/859884
[14] Horizon 2020 project CanPathPro. http://www.canpathpro.eu/
Scientific Contact
Dr. Daniel Weindl
Helmholtz Zentrum München
Institute of Computational Biology
Ingolstädter Landstr. 1, D-85764 Neuherberg (Germany)
e-mail: daniel.weindl [@] helmholtz-muenchen.de
LRZ project ID: pr62li
December 2019